
Abstract
In this paper, the case of a 34-year-old male with sickle cell disease, recurrent episodes of ‘girdle syndrome’, and development of chronic ischaemic colitis is reported. At his last admission to the hospital, he presented with ileus attributed to severe intestinal ischaemia. During his hospitalisation, despite optimal supportive treatment, he developed acute liver failure, possibly as a result of acute intrahepatic cholestasis, a rare but fatal complication of sickle cell disease, and died from sepsis and multiorgan failure.
INTRODUCTION
Acute vaso-occlusive crisis and haemolysis characterise sickle cell disease. Rarely observed, ‘girdle syndrome’ is a situation in which vaso-occlusion occurs in the lungs, liver, and mesentery, and the pain has a characteristic girdle-like distribution. The clinical appearance is nonspecific, with a wide range of manifestations ranging from abdominal pain without significant findings upon physical examination, to ischaemic colitis with a clinical picture of acute abdominal pain and symptoms of ileus and/or colonic perforation. Girdle syndrome is more common in children and, although rare, is more likely to develop during the course of an acute, painful sickle crisis. The authors describe the case of a 34-year-old male who was admitted with girdle syndrome and developed chronic colitis and intrahepatic cholestasis. Acute abdominal pain crises often present with a complicated clinical picture and diagnostic dilemma because it can be difficult to distinguish typical vaso-occlusive pain episodes from their complications and other causes of abdominal pain.
CASE REPORT
A 34-year-old male patient with sickle cell anaemia was admitted to the emergency ward of the author’s institution because of right upper quadrant abdominal pain which started a few weeks before; moreover, the patient noted diarrhoea, worsening of appetite, and fatigue. Physical examination demonstrated jaundice, right upper quadrant tenderness, an enlarged liver, as well as a distended abdomen. He had a history of recurrent pain crises and was submitted to cholecystectomy a few years before because of the presence of gallstones. Laboratory evaluation revealed a white cell count of 14.32×109/L, haemoglobin of 7.60 g/dL, and a platelet count of 515.00×109/L. Serum chemistry showed potassium of 3.8 mmol/L, blood urea nitrogen of 17.0 mg/dL, and creatinine of 0.7 mg/dL. There was a slight elevation of aspartate aminotransferase (49.0 U/L) and lactate dehydrogenase (313.0 U/L), and a marked elevation of total bilirubin (3.6 mg/dL), alkaline phosphatase (410.0 U/L), and γ-glutamyltransferase (563.0 U/L) with normal amylase levels (37.0 U/L). International normalised ratio (INR) was 1.3, prothrombin time was 15 seconds, and activated partial thromboplastin time was 31 seconds. C-reactive protein concentration was 40.30 mg/L. He was found to be a carrier of hepatitis B virus, with undetectable serum hepatitis B virus-DNA levels. He was negative for hepatitis C antigen and positive for IgG antihepatitis A antibodies, and serology tests for cytomegalovirus and herpes simplex virus-1 and 2 were negative. The abdominal ultrasonography showed hepatomegaly with diffuse increases in liver echogenicity without ascites or features of cirrhosis. No gallstones were seen, and the spleen was atrophic, indicating autosplenectomy. A barium enema examination showed thumbprinting in the caecum and ascending colon. A rectal swab culture was obtained and was negative for enteric pathogens. The patient was hospitalised and treated with red blood cell exchange transfusions, analgesics, and intravenous metronidazole and ciprofloxacin. The patient showed improvement and left the hospital after 6 days. Considering the risk of the patient already having a hypercoagulable disorder, they were tested for all five inherited thrombophilias (mutations in the genes that encode protein S, protein C, antithrombin, factor V Leiden, and prothrombin), and for antiphospholipid syndrome, for which no pathological findings were revealed.
In the following months, the patient had repeated hospital admissions for pain crises, abdominal pain, diarrhoea, and weight loss. For every admission he was treated with red blood cell exchange transfusions, intravenous hydration, antibiotics, and analgesics; haemaglobin S levels were maintained at <28%. A lactose-free diet was recommended, but the patient continued to have six to eight loose stools per day. A flexible rectosigmoidoscopy revealed areas of exudate and necrosis in the rectosigmoid colon with multiple blood clots, and biopsies obtained did not show any significant findings. Stool cultures and stool parasitology tests were negative. Abdominal T2-weighted MRI to assess iron overload showed no signs of secondary liver haemochromatosis, with liver iron being 1.1 mg/g (dry liver measurement), but significant liver hypertrophy and upper abdominal varices were suggestive of portal hypertension. CT evaluation of the abdomen revealed bowel distention, thickening of the intestinal wall, mesenteric lymph node enlargement, signs of liver cirrhosis, and presence of ascites. Liver elastography showed an elevated liver stiffness of 13.2 kPa, indicative of liver cirrhosis.
On the patient’s last admission, he had developed serious diarrhoea, of which manifested as >20 loose stools per day, and his abdomen became distended with generalised tenderness and hypoactive bowel sounds. An abdominal radiogram showed elevation of the right hemidiaphragm as a result of the enlarged liver, dilated loops of small bowel but no air-fluid levels (Figure 1). The serum amylase, lipase, alkaline phosphatase, aspartate aminotransferase, and alanine aminotransferase were markedly elevated. A CT scan revealed bowel distention, thickening of the intestinal wall, thumbprinting, and a moderate volume of ascitic fluid (Figure 2). Multiple liver infarcts appeared as circumscribed, peripheral, wedge-shaped areas of decreased attenuation. A peritoneal tap yielded a cloudy fluid which was positive for Escherichia coli. After 2 weeks of being admitted to hospital, the patient began to develop an abrupt onset of jaundice elevation and liver dysfunction with subsequent development of encephalopathy and coagulopathy. Laboratory evaluation revealed a white cell count of 6.34×109/L, haemoglobin of 7.40 g/dL, and a platelet count of 158.00×109/L. Serum chemistry showed potassium of 5.2 mmol/L, blood urea nitrogen of 169.0 mg/dL, and creatinine of 1.2 mg/dL. There was a slight elevation of aspartate aminotransferase (56.0 U/L), alanine transaminase (52.0 U/L), and lactate dehydrogenase (326.0 U/L), as well as a marked elevation of total bilirubin (15.1 mg/dL), alkaline phosphatase (329.0 U/L), and γ-glutamyltransferase (495.0 U/L). C-reactive protein concentration was 29.4 mg/L and INR was 2.4. The patient also developed haematochezia, a tense and tender abdomen, and the absence of bowel sounds. Oral feeding was discontinued, and the patient was placed on parenteral nutrition. The patient required multiple transfusions of packed red blood cells and fresh frozen plasma because of severe reductions in procoagulant and anticoagulant factors, and a dopamine drip for hypotension. Stool examination was negative for Clostridium difficile toxin and blood cultures were negative for cytomegalovirus, HIV, and Yersinia. A blood culture was positive for Klebsiella pneumoniae and therefore he was treated with colistin. Despite prudent clinical care and all applicable therapeutic interventions, the condition of the patient was irreversible, and the patient died as a result of multiorgan failure.
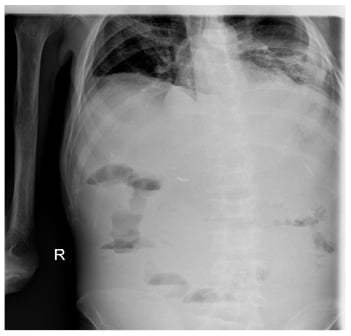
Figure 1: Abdominal X-ray showing elevation of the right haemidiaphragm as a result of the enlarged liver.
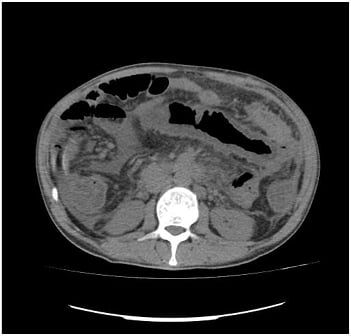
Figure 2: CT evaluation of the abdomen revealed bowel distention, thickening of the intestinal wall, and thumbprinting, indicative of ischaemic bowel disease.
DISCUSSION
Acute abdominal pain is a common symptom in patients with sickle cell disease. It is often difficult for a clinician to deduce whether it is a sickle cell crisis or another intra-abdominal pathology requiring surgical treatment; however, proper diagnosis is mandatory to not only avoid unnecessary laparotomies that can aggravate a sickle cell crisis, but to also provide the appropriate analgesic and supportive treatment.
Acute abdominal pain as a result of a sickle cell crisis is thought to be caused by vaso-occlusions in the microcirculation of the mesentery and the viscera that irritate the peritoneum. In most cases the pain is self-limiting and does not lead to ischaemic colitis.1 A particular clinical entity with rare occurrence is mesenteric syndrome, otherwise known as girdle syndrome, presenting with vaso-occlusions in mesenteric vessels that is often associated with acute thoracic syndrome. The clinical appearance of girdle syndrome is nonspecific, with a wide range of manifestations ranging from abdominal pain without significant findings upon physical examination, to ischaemic colitis with a clinical picture of acute abdominal pain and symptoms of ileus and/or colonic perforation.2 In most cases, the pain is accompanied by haematochezia, but without abdominal tenderness, rebound tenderness, or reductions in intestinal sounds.3 Differential diagnosis of abdominal pain in sickle cell disease includes hepatic or splenic sequestration crisis, hepatic infarctions and acute hepatic crisis, pancreatitis, choledocholithiasis, cholecystitis, appendicitis, and stomach ulcers, occurring at an increased incidence in this population.4-6 A complete blood count is necessary, in conjunction with biochemical control of renal and hepatic function and amylase levels, for the exclusion of these other conditions, as well as blood cross-matching for the onset of transfusion therapy. Artery blood gases and chest X-rays are useful tests to investigate the involvement of the respiratory tract in the crisis. Bowel dilatation and the thumbprint sign are features of ischaemic colitis in the abdominal radiograph and are most visible in the first 24–72 hours.7 Significant for diagnosis is the role of CT imaging, which demonstrates thickening of the intestinal wall with narrowing of the lumen as a result of mucosal oedema. Free air may also be observed in the peritoneum in the event of perforation. Colonoscopy provides the highest diagnostic accuracy, because observation of the haemorrhagic oedematous mucosa and ulcerations characterise ischaemic colitis,8 while microscopic vessel obstructions in the presence of sickle cells are observed in the microscope. However, colonoscopies should be performed with caution because the bowel dilatation reduces blood supply to the intestinal mucosa and may exacerbate ischaemia.9 Conservative treatment with feeding discontinuation, analgesia, transfusion or exchange transfusion therapy, hydration, and potentially antibiotic coverage is typically sufficient, while a laparotomy is required in cases of doubt regarding diagnosis but also for the treatment of complications in persistent vascular obstruction with significant irreversible ischaemia or perforation of the intestine. During a sickle cell crisis, a laparotomy should be avoided because it leads to hypoxia which strengthens the sickling process and halts remission of the symptoms; if necessary, exchange transfusions should be preceded.
Acute intrahepatic cholestasis is the most severe acute clinical manifestation of sickle cell disease concerning the liver and, although rarely, can be fatal. It initially presents as an acute liver crisis with fever and right upper quadrant abdominal pain, and rapidly evolves into acute hepatic failure with coagulation disorders and hepatic encephalopathy. Laboratory findings are characterised by a large increase in bilirubin levels, mainly of its conjugated fraction, due to haemolysis, cholestasis, and renal failure.10 Transaminases also markedly increase and prothrombin time, activated partial thromboplastin time, and INR are prolonged. Treatment is supportive with immediate onset of transfusions or exchange transfusions and administration of fresh frozen plasma for the treatment of coagulation disorders.9 Renal function generally reverts after the treatment of liver insufficiency and chronic dialysis is not required: only temporary dialysis in the acute phase is required. The prognosis is often poor.10,11
This patient had developed chronic ischaemic colitis as a result of repeated episodes of vaso-occlusions concerning the mesenteric vessels. Despite maintaining haemoglobin S levels <28% with exchange transfusion therapy, on his last admission he developed severe colonic ischaemia with the clinical presentation of ileus and multiple liver infarcts leading to acute liver failure, encephalopathy, and coagulopathy (i.e., severe girdle syndrome). The clinical presentation and the laboratory findings were indicative of acute intrahepatic cholestasis. Despite optimal treatment, he died from sepsis and multiorgan failure.
CONCLUSION
This is a teaching case of a patient having received all accessible treatment, but the course of the illness was catastrophic. Sickle cell disease is a benign multisystem disease mandating multidisciplinary management from an experienced medical team. The authors support the introduction of new targeted therapies to significantly reduce these complications from a benign disorder.





