Abstract
Systemic lupus erythematosus (SLE) is a chronic inflammatory autoimmune disorder resulting from autoantibodies produced by B-cell derived plasma cells. Clinical presentation ranges from mild skin rash to multiorgan failure. Regardless of the clinical presentation or severity of the disease, patients with SLE often require life-long treatment. Current treatment recommendations for SLE include hydroxychloroquine, glucocorticoids, immunomodulatory agents, cyclophosphamide, and biologic agents. Despite availability of these agents, the condition of some patients with SLE progressively worsens. With limited treatment options, new and novel therapeutic approaches are needed. Given the active role of B cells in the pathophysiology of SLE, they present an attractive target for therapies evolving in the oncology field. Amongst these, immune effector cell therapies, including chimeric antigen receptor (CAR)-T cell therapy, have proven beneficial in targeting B cells. The eradication of B cells, along with the potential for T cell persistence, has resulted in prolonged remission or stable disease. This review provides an overview of the pathophysiology of SLE; current treatment options, including monoclonal antibodies targeting cluster of differentiation-20 (CD20), CD22, and B cell-activating factor (BAFF); and explores why and how immune effector cell therapies may prove a promising therapeutic option for this patient population, particularly for individuals with refractory disease. Clinical implications from currently approved U.S. Food and Drug Administration (FDA) agents for haematologic malignancies are discussed and provide insight into considerations for applying this therapy to the patient population with SLE in the context of clinical trials.
INTRODUCTION
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with unknown cause and varying clinical presentation, ranging from mild rash to multiorgan failure. SLE predominantly affects females, particularly those of childbearing age. The prevalence is higher in individuals belonging to African American, Asian, or Hispanic ethnic groups compared to those of Caucasian ethnicity.1 The pathogenesis of SLE often involves production of immunogenic forms of DNA, nuclear proteins, and other self-antigens.2 These immunogenic materials provoke the immune system, leading to activation of T cells and B cells and subsequently producing autoantibodies and pathogenic effector cells, such as Th1 and Th17.3 As a consequence, immune complexes between autoantibodies and their cognate antigens are formed and deposited in various tissues causing complement activations. These activated Th1/Th17 cells produce and secrete cytokines and chemokines causing chronic inflammation. If left unchecked, these can lead to severe organ damage and have life-threatening consequences (Figure 1).
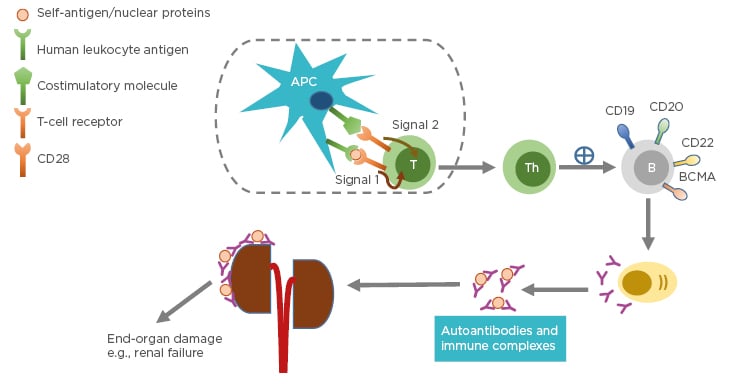
Figure 1: Pathophysiology of systemic lupus erythematosus.
Antigen-presenting cells, such as dendritic cells, uptake self-peptides or nuclear proteins and migrate to draining lymph nodes where they become mature. The mature dendritic cells interact and engage with and activate naïve T cells in the lymph nodes through Signal 1 (human leukocyte antigen: autoantigen complex–TCR interaction) and Signal 2 (co-stimulatory molecule–CD28 interaction). Activated T cells become effector cells i.e., T-helper cells and cytotoxic T-lymphocytes (not shown). The effector cells promote B-cell differentiation into autoantibody-producing plasma cells. Consequently, immune complexes are formed. Deposition of immune complexes at various tissues including the kidneys lead to end-organ damage.
APC: antigen-presenting cell; B: B cell; BCMA: B-cell maturation antigen; CD: cluster of differentiation; T: T cell; Th: T helper cell.
Therapeutic options for the management of SLE have evolved from antimalarial agents such as mepacrine and hydroxychloroquine in the 1950s to immunosuppressive agents including methotrexate, azathioprine, and tacrolimus in the 1960s.4 More recent advances have included the use of targeted therapies such as rituximab, followed by the U.S. Food and Drug Administration (FDA) approval of belimumab, a humanised monoclonal antibody, in 2011.4 Despite availability of these advanced opportunities, it remains essential to explore additional therapeutic options, particularly for individuals with severe presentation of refractory disease. New and novel therapies in the oncology field, specifically immune effector cell therapies, are an attractive option for clinical trials in the patient population with SLE based on their capacity to target specific surface antigens, as well as the potential for persistence which may convey extended therapeutic benefit. This review provides relevant information that could potentially lead to a drug repositioning investigation of chimeric antigen receptor (CAR)-T cell therapies for severe and refractory SLE.
ROLE OF B CELL DEPLETION IN SYSTEMIC LUPUS ERYTHEMATOSUS
B cells are involved in crucial processes of SLE development. The role of B cells in the pathogenesis of SLE includes antigen presentation, T-cell activation, and autoantibody production.5 Depletion of B cells, therefore, is an attractive target for treatment of SLE. In the past decade, there have been several trials designed specifically to address such treatment interventions.6–9 Strategies developed to deplete B cells focussed on identifying targets pertaining to B lineage cells, which include cluster of differentiation-19 (CD19), CD20, CD22, and B cell-activating factor (BAFF).
CD19 is a transmembrane glycoprotein found on B lineage cells, spanning from pre-B cells to plasma cells.10 It is required for normal B cell development and antibody production.11,12 CD19 protein forms a complex with CD21, CD81, and CD225 in the membrane of mature B cells. Together with the B cell antigen receptor, this complex signals the B cell to decrease its threshold for activation by the antigen.13 Thus, CD19 is considered a co-stimulatory molecule for B cell receptor signalling. Deficiency in CD19 expression on B cells leads to hypogammaglobulinaemia.11 Among individuals with SLE, CD19+ B cells are associated with SLE activity, exhibiting elevations in the presence of active versus stable disease. Further, programmed death ligand-1 (PD-L1) has higher expression in patients with SLE compared to healthy controls, and this expression is higher still in patients with active versus stable disease, suggesting a role of PD-L1 in the regulatory mechanisms of B cells.14 Given the activity of CD 19+ B cells with and without PD-L1 expression, they may serve as a therapeutic target in SLE management. There are currently two targeted agents which could potentially be applied as SLE therapy: obexelimab, which targets CD19 and Fcγ receptor IIB (FcγRIIb);15 and CD19-directed CAR, of which there are two currently FDA-approved agents with indication in B cell malignancies. Tisagenlecleucel is indicated for treatment of relapsed or refractory acute lymphoblastic leukaemia and diffuse large B cell lymphoma (DLBCL)16 and axicabtagene ciloleucel is indicated for treatment of relapsed or refractory large B cell lymphoma, including DLBCL, primary mediastinal B cell lymphoma, high grade B cell lymphoma, and DLBCL arising from follicular lymphoma.17
In addition to CD19, other targets for B-cell depletion may include BAFF, CD20, and CD22. CD20 is expressed on all mature B cells including memory B cells and plasma cells. Its function involves B cell differentiation, activation, and proliferation.10 Rituximab, a chimeric monoclonal antibody against CD20 approved for treatment of chronic lymphocytic leukaemia and non-Hodgkin’s lymphoma, efficiently eliminates the majority of B lineage cells; however, it may not deplete the whole memory B cell compartment, leading to disease relapse.18 As a result, clinical outcomes of rituximab in SLE are rather poor.7,8 In addition, rituximab requires repeated administration to achieve and maintain a therapeutic dose. Newer anti-CD20 monoclonal antibodies10 such as obinutuzumab, ofatumumab, veltuzumab, and ocrelizumab, are viable options for SLE, one of which is currently under clinical investigation.19 Another trial studied outcomes of participants with nephrotic syndrome treated with ocrelizumab,20 in which participants without nephrotic syndrome achieved renal response at twice the rate of those with nephrotic syndrome who were treated with ocrelizumab.21 CD22 (also known as Siglec-2), a member of the sialic acid-binding immunoglobulin-like lectins family, serves as an adhesion molecule. It has been shown that blockage of CD22 affects B cell migration in SLE.22 However, epratuzumab, an investigational anti-CD22 monoclonal antibody, did not result in improvements when used in combination with standard therapies for moderate or severely active SLE.9 Lastly, BAFF, also known as B lymphocyte stimulator, is an activating factor for B cell survival and function. Binding of the homotrimer BAFF to the BAFF receptor on naïve and memory B cells enhances B cell survival.23 High levels of soluble BAFF are detected in individuals with autoimmune diseases including SLE,24 making BAFF an attractive target for SLE management. Belimumab is a monoclonal antibody that neutralises BAFF, currently approved for treatment of SLE when used in combination with standard therapies. Unlike monoclonal antibody drugs, CAR-T cells appear to last longer in the systemic circulation, which may extend the period of disease remission.
TARGETING AUTOREACTIVE B-CELLS IN SYSTEMIC LUPUS ERYTHEMATOSUS WITH CAR-T CELLS
The chimeric antigen receptors combine antigen binding domains and signalling domains to facilitate a targeted, receptor-driven response (Figure 2).
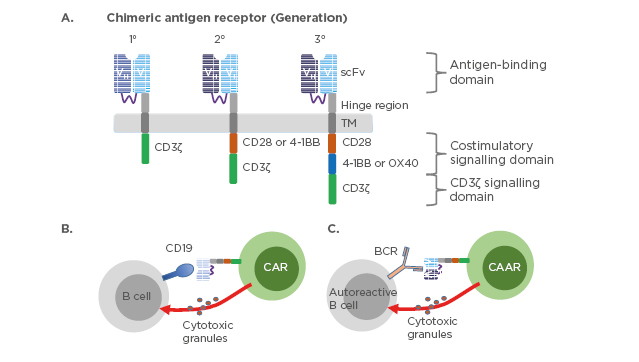
Figure 2: Chimeric antigen receptor constructs and their mechanism of action.
(A) Three generations of chimeric antigen receptors. First-generation CAR are composed of extracellular scFv that are specific to the antigen, transmembrane domain, and cytoplasmic signalling domain CD3ζ. Second-generation CAR incorporate a co-stimulatory domain i.e., CD28 or 4-1BB into the cytoplasmic tail in addition to CD3ζ. Third-generation CAR incorporate two co-stimulatory domains such as CD28 and 4-1BB or OX40. CD19-targeted CAR-T cells (B) or chimeric autoantigen receptor-T cells (C) are genetically engineered to specifically eliminate autoreactive B cells. Direct interaction between CAR-T cells and CD19 expressing B cells or CAAR-T cells and autoreactive B cells expressing B cell receptor recognising autoantigen leads to activation of the CAR/CAAR-T cells to release cytotoxic granules and destroy their targets.
BCR: B cell receptor; CAR: chimeric antigen receptors; CAAR: chimeric autoantigen receptor; scFv: single-chain antibody variable fragment; TM: transmembrane domain.
The success of CAR-T cell therapies in the context of B cell malignancies has led to the exploration of their application to autoimmune conditions, including SLE.25,26 These hypotheses have begun to be evaluated in both murine and human research.27,28 Murine models suggest the potential of CAR-T cells as a potential treatment for SLE, with results showing extended survival and sparing of target organs.27 In these models, purified CD8+ T cells were utilised in place of autoreactive CD4+ T-helper cells, given the potential risk for a disease-enhancing effect in SLE. One study is evaluating the safety and efficacy of CAR-T cells for the treatment of SLE.29 The greatest potential challenge in the application of these therapies to autoimmune conditions is the scarcity of tissue-specific antigens to which the CAR can bind to exert a therapeutic effect.30
In addition, it is hypothesised that the chimeric immune receptors, such as CAR and B cell antigen receptors expressed by T regulatory lymphocytes (Tregs), upon binding with specific T cell conjugates, result in the direct protection of normal cells.30 Among the working hypothesis for the potential role of CAR-T cell therapy is the potential for generating Tregs, which occur at lower frequency in SLE and lead to immune dysfunction. The application of CAR to bind extracellular antigen recognition domains and intracellular hinge domains may be exploited to activate and proliferate Tregs, protecting tissues from a T-lymphocyte attack. There are two mechanisms for expanding Tregs, one involving the use of ex vivo expansion using anti-CD3 or CD28 antibodies, the other involving conversion of conventional T cells to Tregs through the use of transforming growth factor-β alone or in combination with all-trans retinoic acid, rapamycin, or rapamycin alone.31 Once expanded, the CAR engineered Tregs may suppress immune response through several mechanisms including inhibition of antigen-presenting cells, interference with lymphocyte metabolism, secretion of anti-inflammatory cytokines, induction of apoptosis, and cell-to-cell interaction, each of which has the potential to mitigate autoimmune response31 and result in reduced tissue damage. Similarly, studies have demonstrated the capacity of B cell antigen receptor-Tregs to indirectly disrupt B cell function through the suppression of T effector cells.30 It is therefore hypothesised that chimeric autoantibody receptor (CAAR), a chimeric receptor that is designed to express a specific autoantigen, could be used therapeutically for autoantibody-mediated autoimmune diseases, including SLE.30 In addition to their activity against autoreactive B cells, CAAR products may produce persistence and resulting memory cells with potential for reduced off-target toxicity whilst devoid of B cell aplasia and hypogammaglobulinaemia (Figure 2).30
ANTICIPATORY ADVERSE EFFECTS OF CAR-T CELL THERAPIES IN SYSTEMIC LUPUS ERYTHEMATOSUS
Systematic reviews of commercially available CAR-T cell therapies highlight toxicities including cytokine release syndrome (CRS), neurotoxicity, and B cell aplasia.32 CRS is an inflammatory response observed in >70% of patients receiving CAR-T cell therapy and results in fever, hypotension, capillary leak syndrome, and potential for coagulopathies and multiorgan failure.33 CRS most often occurs 1–6 days following infusion, with 95% of cases occurring within the first 12 days post-infusion, though late onset CRS has been observed.34 CRS is also a potential complication associated with the accumulation of transforming growth factor-ß when applied to the autoimmune population.30 CRS is successfully managed with tocilizumab, an IL-6 receptor antagonist. Tocilizumab is a recombinant humanised anti-human IL-6 receptor IgG1k monoclonal antibody and is approved for patients with CAR-T cell-induced severe or life-threatening CRS.35 The Risk Evaluation and Mitigation Strategy (REMS) programme is instituted for both tisagenlecleucel and axicabtagene ciloleucel, as well as tocilizumab, which is a crucial component of the REMS programme.
Immune effector cell-associated neurotoxicity syndrome is believed to be related to fever, high serum IL-6, and high monocyte chemoattractant protein-1 concentrations, and typically occurs within 4–5 days of infusion.36 Symptoms include delirium, headache, and expressive aphasia, with more serious presentations of cerebral oedema and brainstem haemorrhage.36 Management focusses on symptoms and may include corticosteroids to reduce inflammation, levetiracetam as prophylaxis for seizures, and possible use of siltuximab for CRS-related neurotoxicity.
Late effects of CAR-T cell therapy have also been observed. Hypergammaglobulinaemia has been reported, resulting in decreased antibody production with resulting low IgG levels approximately 9 weeks following CAR-T cell infusion and potential persistence for ≥4 years following treatment.37,38 Treatment involves IgG replacement every 3-4 weeks and as needed to maintain therapeutic IgG levels. Monitoring laboratory values to identify critically low IgG is pivotal because of increased risk for infection. B cell aplasia is considered an on-target, off-tumour effect of therapy, resulting in acute and chronic B-cell targeting by CAR-T cells which have demonstrated persistence.39 This persistence, while contributing to B cell aplasia, is also believed to be associated with continued immunosurveillance for malignant cells, thereby resulting in sustained remissions.40
Understanding the known toxicities of therapy is important when considering how such therapies may be applied to the patient population with SLE. Like the current toxicities observed in commercial application of CAR-T cells to haematologic malignancies, CAR products may contribute to cytokine release and on-target toxicities, such as B cell aplasia.32 When applied to patient populations who are autoimmune, the possibility of nonspecific immune suppression may contribute to increased risk of infection or the possibility of undetected tumour growth.31 Newer generation CAR products are demonstrating fewer toxicities in clinical trials.41 Further studies are needed to evaluate the underlying pathophysiologic mechanisms and how these are operationalised when CAR treatment is applied to human subjects with SLE. As these next generation CAR are being applied to a broadening population of patients with haematologic and solid tumour malignancies,42 there is promise for expanding the application of CAR-T cell therapy beyond oncology, with immune-related conditions an optimal therapeutic area.
POTENTIAL SPACE FOR CAR-T CELL THERAPIES IN SYSTEMIC LUPUS ERYTHEMATOSUS
The therapeutic approach and patient populations who are autoimmune, to which CAR-T cell therapies (tisagenlecleucel and axicabtagene ciloleucel) may be safely and successfully applied, has yet to be determined. Given the lack of a current curative option for SLE, consideration might be given to the application of CAR-T cell therapies following trials establishing the safety and efficacy of this approach in humans. Key insights from commercial CAR-T cell therapy approvals suggest that, as well as patients receiving this therapy for oncologic malignancies, individuals with autoimmune conditions might also undergo this treatment. Successes in CD19 directed CAR-T cell therapies in B cell lymphomas have shed some light on CAR-T cell products as a potential treatment modality for severe SLE, including renal (lupus nephritis) and extrarenal disease. Given the current focus of CAR-T cell therapy on CD19 directed B cell malignancies, the same application in SLE may provide an initial target, as is currently being evaluated in a clinical trial (NCT03030976).28 Other potential targets, each of which contributes to T cell proliferation, include the CD3 complex, phosphoinositide-3 kinases, CD44, and IL-2.43
While currently approved CAR-T cell products may have some application for the patient population with SLE, critical questions remain to be answered. First, will this therapy demonstrate therapeutic benefit for this patient population and if so, which products are suitable for which patients with SLE? Data from pharmacological study of second-generation CAR indicates that CD28-based CAR exhibit rapid expansion and boost effector functions, whereas constructs with 4-1BB generate greater persistence and longevity.44 Whether these observations contribute to product selection based on patient characteristics remains to be seen. Experience with tisagenlecleucel and axicabtagene ciloleucel provide insight into clinical applications if and when such therapies are applied to treat SLE; however, a clinical trial is warranted.27
CAAR as opposed to CAR derived products may be most beneficial, given the targeting of autoantibody cells that underlie the pathophysiologic mechanisms of SLE (Figure 2). The broader classification of immune effector cells; genetically modified cells from diverse lineages, including T cells, macrophages, and dendritic cells; and natural killer cells offer unique opportunities to explore various therapeutic targets in the highly heterogeneous SLE presentation.45 Further studies are needed to apply both commercially available and investigational effector cell therapies to the SLE populations to identify which products are most effective in producing a durable response.
CONCLUSION
SLE is an autoimmune disease that can progress to end-organ damage. Standard treatments for SLE may be adequate to control the disease for some patients. Unfortunately, many SLE patients continue to progressively worsen and are refractory to standard of care, requiring novel treatment modalities. Recent approaches include depressing circulating B cells with rituximab, obinutuzumab, ocrelizumab, or belimumab. The efficacy of CD19 directed CAR-T cell therapies in B cell lymphomas may serve as a potential treatment modality for SLE. If results in the SLE population mirror those in individuals with haematologic malignancies, such approaches may require a single treatment that can produce cellular persistence resulting in longitudinal disease control, whilst acknowledging acute and long-term toxicities including CRS, immune effector cell-associated neurotoxicity syndrome, or B cell aplasia. Nonetheless, immune effector cell therapies, including but not limited to CAR-T cell therapies, provide a novel option for evaluating the effect of such therapies on SLE disease response, survival, and quality of life in the context of clinical trials.





