
Abstract
This article will review recent and forthcoming advances in the treatment of thalassaemia. Prognosis of thalassaemia has dramatically improved in the last 50 years with the development of regular and safe blood transfusions and iron chelation. Almost 20 years ago, development of oral chelators, and more recently the improvement in the knowledge and understanding of iron pathophysiology, have led to optimal iron toxicity prevention and treatment. These considerable advancements in medical therapy have transformed transfusion-dependent thalassaemia from a lethal childhood disease to a chronic disease with an open prognosis, even in those individuals over 50 years of age, and with the disease being, in some instances, curable. In the 1980s, the introduction of allogeneic haematopoietic cell transplantation provided the possibility of curing the congenital disease for the first time. More recent developments include an improved understanding of erythropoiesis, which led to the development of new erythroid-stimulating factors effective in thalassaemia, an expansion of donor pull for transplantation, and the approach of the long-term promised gene therapy in clinical practice. Moreover, ongoing trials of gene editing and agents modulating iron metabolism promise new improvements. Today, patients with thalassaemia have several weapons in their therapeutic arsenal and, hopefully, will have much more to come. As usual in medical practice, new advancements provide new challenges for the medical community, and it is the duty of this community to clearly understand the benefits and challenges of any new approach in order to provide the highest clinical benefit to patients.
INTRODUCTION
β-thalassaemias are a clinically heterogenous group of inherited disorders caused by >200 described mutations in the β-globin gene, leading to a decreased or absent production of the β-globin chain. The hallmark of the disease is the imbalance in the α/β-globin chain production, which results in variable grades of ineffective erythropoiesis for apoptosis of late-stage erythroid precursors, chronic haemolytic anaemia, compensatory haemopoietic expansion, hypercoagulability, and increased iron absorption.1 Approximately 1.5% of the global population are carriers for a β-thalassaemia mutation, with traditionally higher prevalence in populations where malaria is or was endemic such as the Middle East, the Mediterranean region, and Southeast Asia;2 however, prevention programmes and migrations have recently contributed to a change in the pattern of thalassaemia distribution worldwide.3 Improved public health measures have prolonged life expectancy of affected individuals in low- and middle-income countries, making β-thalassaemia a significant global health problem.4
Current guidelines have adopted a clinical classification of thalassaemia syndromes based on the magnitude and frequency of transfusion requirements, which are considered to reflect the severity of the disease.5,6 Patients with transfusion-dependent thalassaemia (TDT) present with severe anaemia as early as 6 months of age and require life-long blood transfusion to survive.1 Conversely, in non-transfusion-dependent thalassaemia (NTDT), patients usually maintain haemoglobin (Hb) levels between 7 and 10 g/dL and may require transfusions sporadically. They eventually develop clinically significant iron overload, mainly as a consequence of erythron expansion and increased duodenal iron absorption driven by hepcidin suppression.7 This review aims to provide a summary of the current and emerging treatment strategies for β-thalassaemia.
CURRENT TREATMENTS (STANDARD THERAPY)
Transfusion and Chelation
The aim of red blood cell transfusion in thalassaemia is to restore normal Hb values and to suppress ineffective erythropoiesis, thus attenuating the downstream consequences.1 In chronically transfused patients with TDT, the transfusion-mediated rise in Hb suppresses erythropoiesis and is associated with a rise in hepcidin levels.8 Improvements in blood product administration practices and more effective iron chelation were associated with better survival outcomes and fewer cardiac complications.9 The best course of action to suppress abnormal erythropoiesis, minimise iron overload, guarantee normal growth, and avoid hyperviscosity contemplates a pre-transfusion level between 9 and 10.5 g/dL, with a higher target of 11–12 g/dL for patients with cardiac involvement or with extramedullary pseudotumours.10 Conversely, in NTDT, transfusion may be indicated sporadically or for temporary periods in specific situations;7 clear evidence regarding the most appropriate target of pre-transfusion Hb in NTDT is lacking and some patients, specifically those with the HbE/β-thalassaemia genotype, seem to be able to adapt to lower Hb levels.11 Patients with NTDT placed on transfusion regimens seem to have fewer complications deriving from chronic anaemia and ineffective erythropoiesis (extramedullary haematopoiesis, pulmonary hypertension, thrombotic events);12 on the other hand, given the increased risk of alloimmunisation and particularly of secondary iron overload under chronic transfusion regimens, the decision to commit to a regular transfusion programme must be well thought-out and patients must be evaluated a long time before/after acute episodes.7
In TDT, transfusion iron can accumulate at a rate of 0.3–0.6 mg/kg with a standard chronic transfusion regimen; iron from senescent red blood cells is stored mainly by reticuloendothelial macrophages and rapidly released into the plasma, where transferrin is saturated and labile toxic iron species emerge.13 The most important advancement in iron chelation treatment has been the understanding that iron tissue toxicity is not only related to iron overload but to the presence of toxic iron species in the plasma.14 Therefore, iron chelation therapy (ICT) is administered with the primary goal of removing iron oxidative species emerging in the plasma after transferrin saturation, and the secondary goal of removing formerly deposited iron. An iron overload assessment is thus recommended as soon as 10 units of red blood cells have been transfused, through liver iron concentration (LIC) measurement by MRI or, if not applicable or unavailable due to limited resources, through serum ferritin level measurement. For TDT, a threshold of >1,000 ng/mL is usually an indication for ICT requirement and a value >2,500 ng/mL strongly correlates with cardiac siderosis and endocrinopathy.15
In NTDT, iron overload is related to increased intestinal absorption secondary to suppressed hepcidin. Hepcidin is synthesised in the liver and promotes degradation of ferroportin, the only known cellular iron exporter expressed on macrophages, hepatocytes, and on the basolateral membrane of enterocytes, thus reducing iron absorption.14 Ineffective erythropoiesis and erythroid expansion have been suggested to promote suppression of hepcidin, despite the onset of iron overload, through production of the erythroblast-derived factor erythroferrone.16 This results in a slower rate of iron accumulation in NTDT compared to TDT and in a different distribution, with slower or absent myocardial involvement even in the presence of elevated LIC.17 Guidelines recommend ICT if LIC is >5 mg iron/g dry weight or serum ferritin is >800 ng/mL, although more recent data showed significant iron overload for ferritin values in the 300–800 ng/mL range.18
Three iron chelators are licensed by most regulatory agencies for thalassaemia (deferoxamine [DFO], deferasirox, and deferiprone) and the choice should be carefully evaluated by clinicians. DFO was the first available chelator and is administered parenterally, subcutaneously, or intravenously; it has a short half-life of only 20–30 minutes and needs to be administered as a continuous infusion of 12 hours for 5–7 days per week, thus making it a less attractive choice for non-compliant patients. Despite the introduction of oral iron chelators, DFO is still considered the first-line choice in paediatric patients with TDT under the age of 6 years in Europe and for all patients with TDT in low-income countries due to lower costs. Deferasirox monotherapy at a dose of 20–40 mg/kg once daily proved effective as a first-line therapy for transfusional iron overload in TDT for reducing serum ferritin and LIC and ameliorating myocardial T2* on MRI, even in heavily iron-overloaded patients.19,20 Deferasirox is also the only iron chelator evaluated in a randomised clinical trial in NTDT, showing efficacy in reducing LIC in patients ≥10 years of age with a baseline value of at least 5 mg iron/kg dry weight.21 Deferiprone is an oral (administration 3 times per day) absorbed iron chelator licensed in most countries as a second-line therapy for iron overload in patients with TDT >6 years of age. Its efficacy in LIC reduction has also largely been demonstrated.22 Sporadic severe cases of granulocytopenia have been reported. Notably, a combination therapy of DFO and deferiprone proved effective in the management of cardiac overload, by ameliorating cardiac T2* on MRI and left ventricular ejection fraction.23
Allogeneic Haemopoietic Cell Transplantation
The rationale for haematopoietic cell transplantation (HCT) in thalassaemia is to replace ineffective endogenous erythropoiesis and to correct the phenotypic expression of the disease, sparing patients from life-long transfusion treatment and long-term complications.24 HCT is, so far, the only consolidated approach with a curative potential in TDT. Transplantation in TDT is now performed worldwide with excellent results.25-28 The latest report from The European Society for Blood and Marrow Transplantation (EBMT) Registry showed a global overall survival (OS) and event-free survival (EFS) of 88% and 81%, respectively, with best results obtained in patients ≤14 years of age (OS and EFS of 90% and 83%, respectively).29 Current recommendations identify young patients with TDT, before development of iron-related organ damage, as the ideal candidates for HCT;30 adults can also be offered this strategy in the setting of dedicated programmes, provided that they have been well-chelated since infancy. According to the EBMT Registry data, better outcomes are still obtained from transplants with human leukocyte antigen (HLA)-matched siblings, with an OS of 91% and EFS of 83%.
The possibility of finding an HLA-matched sibling donor ranges from 60 to 70% in countries where families are larger and an average of 25–30% in western countries,31 highlighting the need for alternatives. In the era of high-resolution HLA typing, transplantation from matched unrelated donors in TDT is considered feasible and effective. Transplantation from alternative donors has resulted in conflicting results so far. Primary graft failure appears as the major complication of unrelated cord blood unit transplants, while the need to abate the risk of graft-versus-host disease has prompted the search for effective modalities of ex vivo or in vivo T-cell depletion with grafts from haploidentical donors. Recent reports of alternative HCT donors are summarised in Table 1.32-38
Table 1: Haematopoietic stem cell transplantation from alternative donors in patients with thalassaemia.
aGVHD: Acute graft-versus-host disease; ATG: anti-thymocyte globulin; Az: azathioprine; Bu: busulfan; cGVHD: chronic graft-versus-host disease; CsA: cyclosporin A; Cy: cyclophosphamide; Dxm: dexamethasone; Flu: fludarabine; GF: graft failure; GVHD: graft-versus-host disease; HLA: human leukocyte antigen; Hu: hydroxyurea; MAC: myeloablative conditioning; MMF: mycophenolate mofetil; MMUD: mismatched unrelated donor; MP: methylprednisolone; MTX : methotrexate; MUD: matched unrelated donor; N/A: not applicable; NR: not reported; OS: overall survival; PT-Cy: post-transplant cyclophosphamide; Rit: rituximab; TFS: thalassaemia-free survival; TT: thiotepa; UCB: umbilical cord blood; Vel: velcade.
Erythroid Maturating Agents
Members of the TGF-β superfamily of ligands, including several bone morphogenetic proteins, growth-differentiation factors, and activins, have been known to be inhibitors of late-stage erythropoiesis. Their complex interaction with erythroid precursors was recently reviewed.39 Activin receptor ligand traps (sotarcept and luspatercept) are fusion proteins that act as extracellular traps for a wide range of ligands of the TGF-β superfamily, thereby preventing receptor binding and stimulation of downstream signalling. This approach proved capable of correcting anaemia in an erythropoietin (EPO)-independent fashion in mouse models of ineffective erythropoiesis, thus raising interest surrounding its possible clinical application in thalassaemia or myelodysplastic syndromes.40 The specific pathway inhibited by these drugs is still a matter of debate, with growth-differentiation factor-11 signalling primarily hypothesised as the major target;40 however, this was recently contradicted by further experiments on murine models.41
Luspatercept (ACE-536) is an activin Type IIA ligand trap, binding to several members of the TGF-β superfamily. It has recently been approved by the U.S. Food and Drug Administration (FDA) and by the European Medicines Agency (EMA)42 for the treatment of anaemia in TDT and EPO-refractory myelodysplastic syndrome with ring sideroblasts. Results from a Phase II trial43 led to the initiation of the randomised, Phase III BELIEVE clinical trial44 of luspatercept in 336 adult patients with TDT in 15 countries. The percentage of patients who had a minimum reduction of 33% of transfusion burden was higher in the luspatercept arm than the placebo arm for fixed predefined time periods (from baseline to Weeks 13–24: 21.4% versus 4.5%; from baseline to Weeks 37–48: 19.6% versus 3.6%, respectively) and during any 12-week period (70.5% versus 29.5%, respectively). Subgroup analysis revealed that the magnitude and rapidity of response might be lower in patients with a β0/β0 genotype than in patients with a non-β0/β0 genotype. Adverse events were mainly Grade 1 and 2 and consisted of bone, back, and musculoskeletal pain, headache, myalgia, arthralgia, and injection site pain. However, in this randomised trial, eight thromboembolic events (deep vein thrombosis, ischaemic stroke, superficial thrombophlebitis, pulmonary embolism) were reported, all in patients who had previously undergone splenectomy.
Given the central role of the JAK2/STAT5-signalling pathway in EPO/EPO receptor interaction on erythroblasts,45 JAK2 inhibition was tested as a potential mechanism for reversing ineffective erythropoiesis. Despite initial promising results,46 when ruxolitinib was tested in a Phase IIa study for patients with TDT with spleen enlargement, no significant Hb increase from baseline was observed nor significant changes in total body iron concentration.47
Gene Therapy: Gene Insertion Approaches
While HCT has long-been considered the only curative treatment for thalassaemias, its applicability is hampered by the availability of a full-matched, HLA-identical donor, by the risk of immunological complications, and by the need for long-term immunosuppression. The goal of current gene therapy is to provide a curative option for patients lacking HLA-identical siblings by inducing the production of the β-globin or γ-globin, thus improving the α/non-α globin ratio, decreasing the deposition of insoluble α-hemichromes, and correcting ineffective erythropoiesis. This can be achieved through two different major platforms: gene insertion and gene editing.48
The first approved gene therapy product approved in Europe for the treatment of patients with TDT aged ≥12 years with a non-β0/β0 genotype, candidates for HCT but lacking an HLA-identical sibling, is LentiGlobin BB305,49 approved in 2019, and belongs to the first category; this consists of inserting a lentiviral vector containing the β-globin-producing genes together with their regulatory machinery (promoter, enhancer, and part of the locus control region) into previously collected autologous haematopoietic peripheral blood stem cells (PBSC) and later infusing these genetically modified PBSCs back into the patient after proper myeloablation to facilitate engraftment.50 The β-globin-producing sequences are placed under the control of an erythroid promoter, so that they can only be transcribed in erythroid precursors. However, this process remains fairly uncontrolled and, despite the optimisation of the more recent lentiviral vectors, occurs ‘semi-randomly’, potentially retaining a risk of clonal proliferation stimulation.52 For this reason, the FDA and EMA require a total follow-up of 15 years for all patients treated with gene therapies.
LentiGlobin BB305 was tested in two Phase I/II trials on a total of 22 patients aged 12–35 years with TDT of any genotype.52 The conditioning regimen consisted of myeloablative doses of busulfan modulated by pharmacokinetic (PK) analysis. All patients attained engraftment and 10% developed veno-occlusive disease and required defibrotide treatment. The vector copy number of infused products ranged from 0.3 to 1.5 in the first trial and from 0.8 to 2.1 in the second trial. The gene-insertion-derived HbA, separately identified by liquid chromatography due to an amino-acid substitution (HbAT87Q), was produced at an average of 6 g/dL in non-β0β0 genotypes, granting transfusion independence in all but one patient. Subsequent efforts have focused on improving the transduction process, elevating the vector copy number of the final product to a median of three copies per double-stranded DNA. The results of the Phase III Northstar-2 and Northstar-3 trials show a stable Hb ≥11 g/dL in 91% of patients with non-β0β0 genotype,53 and 3 out of 4 β0β0 patients followed-up for ≥6 months having stopped transfusions.54 The commercialisation of LentiGlobin for TDT has recently been suspended after two myeloid neoplasms were diagnosed in patients with treated sickle cell disease during follow-up.55 These events will need to be clarified before proceeding with a large clinical application.
In Italy, nine patients were treated with the GLOBE lentiviral vector-transduced autologous haematopoietic PBSCs, with intra-bone administration to avoid trapping into filter organs and allow for better and earlier haematopoietic recovery.56 The most significant gene therapy clinical trials conducted so far in TDT are listed in table 2.52-57
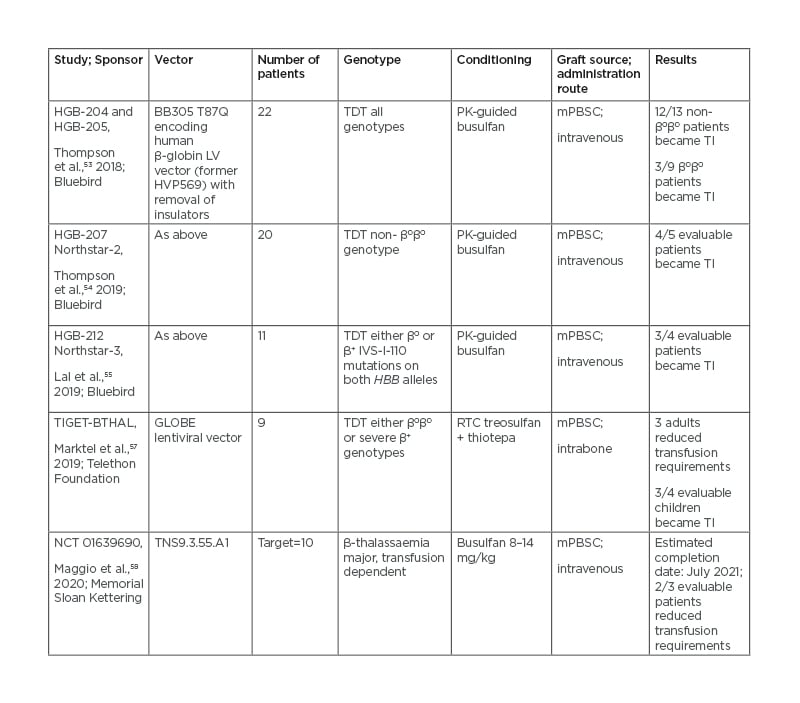
Table 2: Gene insertion approaches in clinical trials.
mPBSC: Mobilised peripheral blood stem cells; PK: pharmacokinetics; RTC: reduced toxicity conditioning; TI: transfusion independent; TDT: transfusion-dependent thalassaemia.
Although gene therapy overcomes the risk of graft-versus-host disease and the need for a suitable donor, there are several challenges still to be faced. 1) Busulfan-based myeloablative conditioning is not devoid of toxicity and some attempts have been made to minimise it, such as dose-tailoring based on PK54 or the use of the less-toxic thiotepa and treosulfan.56 2) Long-term follow up is still not available and the number of treated patients so far is not enough to have a complete safety profile. 3) A large number (≥10–15×106 CD34+/kg) of stem cells is required to account for stem cell losses during manufacturing, demanding for upfront plerixafor.58 4) Unlike stem cell transplantation, where the β-globin defect correction occurs in a pan-cellular fashion (e.g., in every donor-derived stem cell), the number of gene insertions in PBSC can vary depending on the efficiency of manufacturing processes, leading to a heterocellular correction and strongly demanding for an adequate characterisation of the product prior to infusion, at least in terms of viability, purity, and efficiency of the transduction process.48 5) In order to be potentially manufactured on a large scale, costs and quality of life should be superimposable compared to alternative-donor HCT, and a socioeconomic advantage over the costs of life-long transfusions and chelation should be demonstrated.
FUTURE TREATMENTS
Gene Therapy: Gene Editing Approaches
The gene editing approach exploits the possibility to precisely cut human DNA at specific locations by engineered nucleases, such as zinc-finger nucleases and CRISPR/Cas9.60 By doing so, they can either act on specific erythroid enhancer regions regulating the switch from the γ-globin genes to the β-globin genes, or recreate the mutations seen in hereditary persistence of fetal Hb, thus incrementing the production of fetal Hb. This method retains the advantage of a higher precision and efficiency in DNA edits and of more affordable costs.49
Recently, the results of the first patient treated with a CRISPR/Cas9 gene-editing product, targeting the enhancer of the BCL11A gene on chromosome 2, were presented (CTX001 product): the patient (β0/IVS-1-110 genotype) was transfusion-independent at 12-months follow-up and 99% of erythrocytes expressed high levels of fetal Hb.60
More attempts are ongoing with CRISPR/Cas12 products performing edits on the BCL11A binding site on the HBB gene; this process demands extreme precision in order to prevent disruption of endogenous globin production.61 The main limitation lies in the risk to create unintended edits in the genome (‘off-target effects’), although this is not necessarily of clinical significance;62 additionally, chromosomal rearrangements or instability can occur and require serial monitoring.63
Iron Metabolism Modifying Agents (Minihepcidines, Ferroportin Inhibitors)
The hepcidin/ferroportin axis is a major regulator of erythropoiesis; ineffective erythropoiesis with low or inappropriately normal hepcidin levels and subsequent iron overload are hallmarks of the so-called ‘iron-loading anaemias’, with NTDT as a prototype,64 thus the possibility to therapeutically target this axis has recently gained some interest.
Studies on β-thalassaemia in mouse models demonstrated that transgenic overexpression of hepcidin or genetic disruption of hepcidin regulators can result in the prevention of iron overload and enhance haemopoiesis.65 Biosynthesis of full-length hepcidins is inefficient and they are rapidly cleared by the kidneys; however, these limitations were somehow overcome by the development of minihepcidins (MH): short, engineered peptides with an increased half-life and potency, able to mimic the iron-restrictive effect of endogenous hepcidin.66 In young Hbbth3/+ mice, serving as models for NTDT, MH ameliorated anaemia, ineffective erythropoiesis, splenomegaly, and iron overload. In older mice, the administration of MH together with deferiprone did not modify its beneficial effect on iron overload.67 In TDT mice models, MH in combination with chronic red blood cell transfusions further improved ineffective erythropoiesis, splenomegaly, and iron overload. Based on these results, multiple trials were started to assess efficacy of MH in the clinical setting. In a Phase II study, LJPC-401 was tested for the treatment of myocardial iron overload in patients with TDT but the trial was prematurely terminated because of absence of efficacy.68
Another approach to modulate iron metabolism is through ferroportin inhibitors. Among these, VIT-2763 is a small, oral compound that competes with hepcidin for ferroportin binding. In the Hbbth3/+ mice, it ameliorated ineffective erythropoiesis and the dysregulated iron homeostasis; it also corrected the proportion of myeloid precursors in Hbbth3/+ mice spleens.69 A Phase I, double-blind, dose-escalating study was performed in order to assess for safety, tolerability, PK, and pharmacodynamic properties of VIT-2763 in healthy volunteers: no serious events led to drug discontinuation and most of the adverse events in the single and multiple escalating dose cohorts were drug-unrelated; a temporary decrease in mean serum iron and mean transferrin saturation were observed.70 A Phase II, randomised, double-blind trial assessing efficacy, safety, and tolerability of VIT-2763 is currently recruiting in multiple sites.71
CONCLUSION
The last 50 years have witnessed dramatic improvements in thalassaemia understanding and patient care. These improvements have built a series of previously unimaginable therapeutic opportunities for patients with thalassaemia, with many more on the way. All of this was made possible by a synergy between the various fields of biological and clinical research, which have mutually reinforced one another to lead to shared success. Figure 1 reports the milestones of therapeutic progress in β-thalassaemia.
Figure 1: Milestones of therapeutic advances in β-thalassaemia, revealing continuous improvement during recent decades.
Having access to many therapeutic opportunities is undoubtedly beneficial for patients, yet it can also lead to problems when choosing treatment. As opportunities have grown, the cost of optimal therapies has increased dramatically, and so has the demand for a better selection of the appropriate sequence of treatments in terms of cost/benefit ratio. When compared to HCT (the only other available curative option), gene therapy results in, on average, an additional 300,000–400,000 EUR/patient, justified by the high costs of the viral vector and preparation procedures. On the other hand, as competition between different suppliers grows and follow-up monitoring becomes less stringent, the whole procedure should become more affordable;72 nonetheless, requirements in terms of professional skills, quality efficacy, and regulatory compliance still make it an unattractive approach in low-income countries. Traditional treatments, on the other hand, are not devoid of costs, estimated around 30,000 EUR/year/patient for TDT in Italy,73 making HCT at younger ages, when feasible, probably the most appropriate choice in the circumstance, even in terms of resource application. In conclusion, in order for the points discussed in this article to become common practice, an effort must be made to progress, consistent with resource availability and still crucially impacting the real opportunities for benefiting from these advances in the real world.





