
INTRODUCTION
During adulthood and embryogenesis, fate decisions of haematopoietic stem and progenitor cells (HSPC), such as specification, self-renewal, and differentiation, are tightly regulated by their neighbouring niche cells.1 Moreover, distinct types of niches supply differential cues to direct alternative cell fates for HSPC.2,3 Currently however, the intrinsic mechanisms balancing HSPC response obliqueness to microenvironmental signals are unknown.
Fli-1 is an E26 transformation specific transcription factor expressed by vascular beds and haematopoietic lineages. Fli-1 belongs to the family of ‘heptad factors’ which are hypothesised to specify and sustain a haematopoietic cell fate.4,5 While Fli-1 overexpression is linked to leukaemia,6 the functional role Fli-1 plays in HSPC specification and maintenance remains undefined.
METHOD AND RESULTS
The authors showed that inducible global deletion of Fli-1 using a Cre/lox model (Fli-1ROSAΔ ) results in a rapid thrombocytopenia-associated mortality. Transplantation of Fli-1ROSAΔ bone marrow cells into wild-type (WT) recipients, to exclude vascular-mediated defects, followed by induction of Fli-1 deletion resulted in the same phenotype. In a set of modulated competitive transplantation experiments (differential induction time points pre or post-transplant), the authors observed defective ability of Fli-1ROSAΔ HSPC to lodge, engraft, and to sustain haematopoiesis post-repopulation. Fli-1-deficient HSPC exhibit reduced quiescence, a hallmark of stemness, and display enhanced apoptosis. Thus, Fli-1 is essential for previously unrecognised cell-autonomous HSPC functions.
To determine whether Fli-1 deletion abrogates haematopoietic specification, Fli-1 was conditionally deleted using a developmental haematopoietic Cre/lox model (Fli-1Vav-1Δ) which resulted in premature mortality. Reduced presence of embryonic Fli-1Vav-1Δ liver HSPC was observed at e12.5. Moreover, using a developmental endothelial (CDH5)-Cre/lox model (Fli-1CDH5Δ), the authors observed that reduced numbers of haematopoietic cells were still detected in the aorta-gonad-mesonephros (AGM) region. Two in vitro co-culture systems were also applied to study Fli-1 in the endothelial-to-haematopoietic transition. First, isolated haemogenic endothelial cells from WT and Fli-1ROSAΔ embryos were cocultured with AGM-derived vascular niche.7 Haemogenic endothelial cells isolated from Fli-1ROSAΔ AGM were still able to convert to CD45+ cells, however these cells did not expand on a vascular niche. Secondly, an endothelial-to-haematopoietic reprogramming system was implemented in which isolated lung endothelial cells were virally introduced with DOX inducible FosB, Gfi1, Runx1, and Spi1 (FGRS) factors and co-cultured with vascular niche cells.8,9 Both WT and Fli-1ROSAΔ endothelial cells were able to acquire a haemogenic-like state resulting in a final capacity to convert into haematopoietic cells. Again, Fli-1ROSAΔ cells displayed fewer numbers of CD45+ cells at the end point, presumably due to impaired interaction with the vascular niche. Induction of Fli-1 deletion in vitro in adult HSPC revealed loss of dependency on vascular niche inductive signals, as no additive expansion effect was observed for Fli-1ROSAΔ HSPC in the presence of a vascular niche. Hence, Fli-1 is not essential for haematopoietic specification but rather essential for HSPC expansion and self-renewal.
Differential RNA-seq analysis combined with epigenetic studies of expanding WT and Fli-1ROSAΔ HSPC revealed dysregulation of Fli-1-controlled pathways involved in transduction of microenvironmental signals for self-renewal. Focusing on HSPC-vascular niche interaction, this study observed dysregulation in Notch and VEGF pathway elements that mediate an HSPC-vascular niche crosstalk.10
CONCLUSION
Decrypting the mechanism(s) by which Fli-1 orchestrates HSPC self-renewal may promote an improved expansion protocol of human HSPC pre-transplantation and provide additional insights for microenvironmental sensing by Fli-1-dependent normal and leukaemic cells (Figure 1).
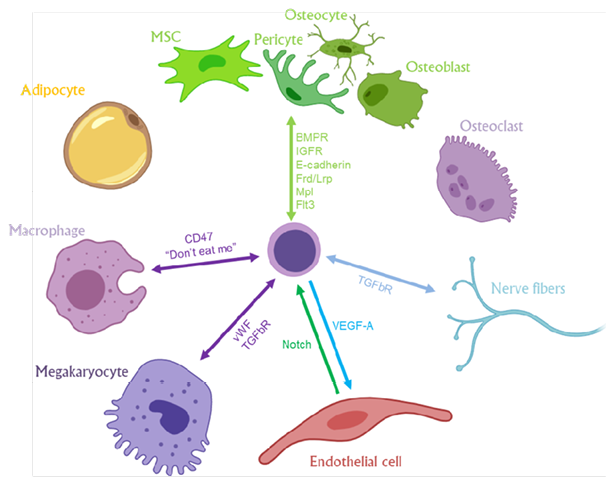
Figure 1: A summary model presenting Fli-1-regulated sensory elements that may mediate the interaction and crosstalk between haematopoietic stem and progenitor cells and their diverse neighbouring niche cells in the bone marrow microenvironment.
MSC: mesenchymal stromal cell.





