Meeting Summary
This symposium was held as part of the 28th International Annual Congress of the World Muscle Society (WMS), held in Charleston, South Carolina, USA. Speakers reviewed the natural history of Becker muscular dystrophy (Becker), outlined the top line, 12-month data from the ARCH open label study of investigational agent EDG-5506, and aimed to put those results into clinical context.
Becker is a serious rare disease with significant physical, emotional, financial, and social impact on the affected individuals and their caregivers. Once function begins to decline, males affected by the progressive X-linked genetic disorder continue to irreversibly lose muscle, ultimately leading to the loss of ambulatory and cardiopulmonary function.
After discussing the aetiology of the condition, Erik Niks, Paediatric and Adult Neurologist, Leiden University Medical Center (LUMC), the Netherlands, presented the findings of natural history studies. They showed that while the age at which decline begins varies, once it does start, patients tend to experience a consistent decline in function equivalent to around 1.2–1.3 North Star Ambulatory Assessment (NSAA) points each year. This finding, combined with data on using MRI as a biomarker of disease progression, provides an evidence-based framework for clinical trial design, he argued. Sam Collins, Vice President of clinical development, Edgewise Therapeutics, Boulder, Colorado, USA, then presented topline 12-month data from the ARCH study. It found that EDG-5506 was well tolerated, and, importantly, recorded the stabilisation of functional assessments, including the NSAA, with a trend towards improvement, as well as rapid, sustained, and significant decreases in biomarkers of progression, including those related to muscle damage. Putting the ARCH study data into context, Barry Byrne, Director of the Health Center for Advanced Therapeutics and Powell Gene Therapy Center, University of Florida (UF), Gainesville, USA, explained exactly how declining NSAA status translated into life-altering function loss. Stabilising function, or even reducing the speed of decline, was an important goal for patients, he said, adding that meeting it could help to address significant unmet medical need.
Background
First described by German physician Peter Emil Becker in the 1950s, Becker is an X-linked genetic recessive disorder.1 Like Duchenne muscular dystrophy (Duchenne), it is caused by a mutation in the gene that produces dystrophin, a protein that links the sarcolemma to the cytoskeleton in the muscle fibres.2 The 79 exons of the dystrophin gene include several internal promoters that give rise to shorter isoforms, and a central region that encodes for the binding domain of neuronal nitric oxide synthase. In general, in-frame mutations lead to Becker, while out of frame mutations lead to a Duchenne phenotype, with both conditions being part of a spectrum of dystrophinopathies. While in Duchenne exon deletions tend to completely block dystrophin production, in Becker the proteins are usually shortened, and only partially functional.2
“The dystrophin itself is located at the periphery of all the muscle fibres. Its expression is clearly reduced in patients with Becker, and nearly absent in Duchenne,” said Niks, describing Becker as being “on the same spectrum” as Duchenne, which has an earlier onset and faster progression.2 The dystrophin protein can be quantified using Western blot analysis, “but it is important to realise that levels of dystrophin are very variable both in healthy controls, as well as in patients with Becker,” said Niks.3 The absence or reduction of dystrophin results in a dystrophic pattern on muscle biopsy, with increased fibrotic tissue and increased fatty replacements being clearly visible on MRI.3
The natural history of Becker is highly variable between individuals.4 Patients will usually experience slowly progressing proximal and paraspinal weakness, and a mixture of progressive muscle hypertrophy and atrophy.4,5 Many, but not all, males with Becker tend to remain ambulant beyond the age of 16 years.5 Muscle pain and cramps are common, and cardiomyopathy can occur later in life.4 Clinicians employ a number of approaches to measure disease activity and progression, including the NSAA, which has been validated for use in clinical trials.6 It consists of 17 items related to everyday activities, such as walking, rising from a chair, and climbing stairs. Each item is scored from 0–2, depending on whether the patient is unable to perform the task (0 points), can perform the task with difficulty or assistance (1 point), or can perform the task independently (2 points). It yields a maximum score of 34 points, with the lowest scores representing the highest levels of function loss.6
Natural History
An Italian natural history study, published in 2016, followed 69 patients between the ages of 6–69 years for 12 months.7 A total of 28 participants had a deletion ending on exon 45, 10 had a deletion of only exon 48 (del 48), 10 had a deletion ending on exon 51 (del x-51), and 21 were classified as ‘other’. The mean NSAA at baseline was 25.3, with a standard deviation (SD) of 10.8. The mean change at 12 months was -0.9, with an SD of 1.6 (p<0.001). Interestingly, participants with the del 48 and del x-51 mutation recorded almost maximum NSAA scores at baseline (del 48: NSAA of 33.9 and SD of 0.32; del x-51: NSAA of 33.7 and SD of 0.95) and little decline over the follow-up period (del 48: -0.3 and SD of 0.50; del x-51: 0.0 and SD of 0.00). In contrast, those with the deletion ending on exon 45 had lower NSAA at baseline (NSAA of 20.9 and SD of 11.10), and a higher degree of change at 12 months (-1.3 and SD of 1.70).7
Niks’ team has recently concluded a similar study in the Netherlands. Presenting the data ahead of publication, he said 36 patients with Becker aged between 18–67 years were followed up for 3 years, with annual functional assessments, and an optional muscle MRI at visits one and three. Eleven participants had one of the most common Becker mutations,8 deletions of exons 45–47, and based on the mutation, the neuronal nitric oxide synthase binding site was predicted to be involved in 21 cases. The study recorded no clear differences in decline between these two subpopulations, leading Niks to say there was “still much to learn about the genotype/phenotype correlations.”
“While we saw patients with a maximum score at baseline who remained basically stable for the entire course of the study, there was a quite consistent decline in the NSAA score over 3 years, mainly in those who had a baseline value between 10 and 32,” he said. The 10 metre walk/run velocity results showed higher variability, an absent genotype/phenotype correlation, and a much less consistent decline, even over 3 years of follow-up.
MRI as a Biomarker in Becker Muscular Dystrophy
Fat fraction (FF), as measured by MRI, may be a useful biomarker of disease progression for use in clinical trials. However, it differs both between and within muscles.9 As part of a study by Niks’ group, MRI and clinical assessments were performed at baseline (n=24) and 24 months (n=20). They used the Dixon method to quantify FF.9
Researchers manually delineated images of 12 thigh and seven lower leg muscles into 23 slices each, calculating FF values for the three central slices, as well as the whole muscle. The functional tests NSAA, 6-Minute Walk Test (6MWT), and 10-metre run velocity were also conducted. “We noticed quite a consistent increase in FF over 24 months, with the median whole muscle FF increasing by between 0.2% and 2.6%, depending on the muscle,” said Niks, adding that increases occurred both in muscles with a relatively low, and higher FF, at baseline. “It could be a biomarker that captures disease progression across the range of the phenotypes,” he added.9
In terms of functional assessment results over the 24 months, the median change in NSAA was -2.5 points (range: -12.00–1.00; p=0.002), and, in 10-metre run velocity, -0.22 m/s (range: -1.40–0.25; p=0.014). This was consistent with figures recorded in previous natural history studies, Niks explained. Next, the researchers conducted a stepwise analysis of the full dataset to identify the optimal MRI biomarker. “Whole thigh FF was very sensitive, reproducible, and also relatively practical to put into an MRI protocol, with standardised response means of around one,” said Niks.9
Summing up, he said: “High clinical variability and slow disease progression does not facilitate the conduction of clinical trials […] but we are able to capture changes in motor function when following these patients for 12 months or longer. When you select subpopulations, for example, those with a baseline NSAA of 10 to 32, you are able to capture progression over the duration of a clinical trial, with a feasible number of patients.”
Targeting Contraction-Induced Muscle Damage
Biomarkers of muscle damage can also be useful in clinical trials. Contraction damages muscles in everyone, but in Duchenne and Becker, the lack of dystrophin results in excessive contraction-induced damage, leading to excessive degeneration and muscle membrane leak.10 This is evidenced by elevated levels of creatine kinase in the blood of people with Becker and Duchenne.11 Data comparing levels of fast skeletal muscle troponin I and slow skeletal muscle troponin I in the blood of healthy controls with people with Becker and Duchenne have also demonstrated that the muscle damage primarily affects the fast muscle fibres in those living with the conditions.11
Explaining how this data has informed the development of the investigational agent EDG-5506, Collins said: “As the sequence of damage continues, it exhausts the regenerative processes, and fat infiltration and fibrosis occur; but if you could protect the muscle against that excessive degeneration, you could help preserve function and, ultimately, muscle function.”
ARCH Study of EDG-5506
Collins went on to outline the 12-month top line results from the ARCH study of EDG-5506, an orally administered, allosteric, selective, fast myofiber (Type II) myosin inhibitor (unpublished data, presented at WMS 2023).12 The compound has been designed to protect the muscle fibres against contraction-induced damage, and the associated loss of function.13
The open-label, single-centre trial enrolled 12 males with Becker aged between 18–55 years, with a dystrophin mutation, a Becker phenotype, and the ability to complete a 100 m timed test (unpublished data, presented at WMS 2023).12 “We were looking at the safety and tolerability of EDG-5506, but we could also measure biomarkers of muscle damage, and carry out functional assessments throughout the course of the study,” said Collins, adding that ARCH had now been extended for an additional year.
After screening, participants were treated with a daily 10 mg dose of EDG-5506, escalating to 15 mg after 2 months, and 20 mg after a further 4 months.12 At baseline, participants had significant functional impairment, with a median NSAA of 15.5 (range: 4–31),12 showing they were in the functional decline phase of disease course, as defined by previous natural history data (unpublished data, presented at WMS 2023).7 They had decreased muscle mass and substantial elevation of creatine kinase, a biomarker of muscle injury (Table 1).
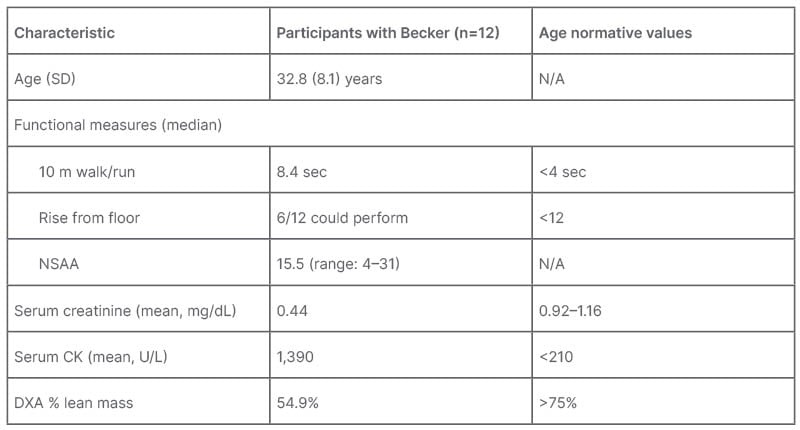
Table 1: Baseline characteristics of ARCH study participants.
Becker: Becker muscle dystrophy; CK: creatinine kinase; DXA: dual energy X‐ray absorptiometry; N/A: not applicable; NSAA: North Star Ambulatory Assessment; SD: standard deviation.
The compound was well tolerated at all doses, and no dose reductions due to adverse events (AE), treatment discontinuations, or serious AEs were reported.12 Over the 12 months, the most common AEs were dizziness, COVID-19, and arthralgia, with each occurring in four (33%) participants. There was no increase in the incidence of these AEs as dose increased (unpublished data, presented at WMS 2023).12
EDG-5506 led to a sustained decrease in biomarkers of muscle damage over the 12 months of treatment. The study recorded an overall 37% (p=0.001) reduction in serum creatine kinase, and a 79% (p<0.0001) reduction in fast skeletal muscle troponin I.12 The observation that biomarkers of muscle damage decreased during the trial suggests treatment decreased contraction-induced injury, said Collins.
Over the course of the 12 months, 75% of NSAA scores either stabilised or improved, and there was a mean 0.4 point improvement (unpublished data, presented at WMS 2023),12 compared with the -1.2 point decline predicted by natural history data (Figure 1).7,9 “It is certainly starting to move away from what you might expect to see in Becker individuals over that kind of time frame,” said Collins, adding that the data pointed to a functional benefit of improving contraction-induced injury.
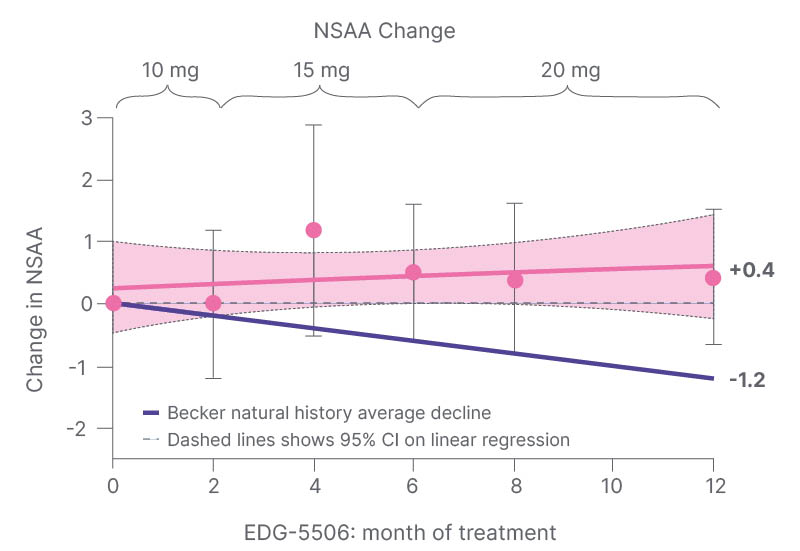
Figure 1: Improvement of North Star Ambulatory Assessment during 12 months of treatment with EDG-5506.
Means±95% CI. Source: data on file.
Natural history based on data presented by Bello et al.7 and van de Velde NM et al.9
Becker: Becker muscle dystrophy; CI: confidence interval; NSAA: North Star Ambulatory Assessment.
In addition, there was no decline from baseline in 100m timed test velocity.12 “We were very pleased to see this. These patients decline over time, so stability in a functional test over a year is a good outcome,” explained Collins. A positive downward trend in patient-reported pain scores was also observed during 12 months of treatment, though it is important to note that the ARCH study was not placebo-controlled.12 “In future studies, we are interested in collecting more data about pain, because it can have a big impact on patients’ day-to-day lives,”14 he went on. “In conclusion, EDG-5506 was well tolerated at all doses; demonstrated rapid, sustained, and significant decreases in multiple biomarkers of disease progression; and resulted in stabilised functional assessments, with a trend towards improvement.”12 Maximal biomarker response was observed at the lowest administered dose of 10 mg (unpublished data, presented at WMS 2023).12
Edgewise (Boulder, Colorado, USA) is now recruiting for GRAND CANYON, a global, 18-month pivotal trial to evaluate the safety and efficacy of EDG-5506 in adults living with Becker.15 Participants will be ambulatory males aged between 18–50 years, with a genetic diagnosis of Becker, and an NSAA of between 5–32. The primary endpoint will be change in NSAA at 18 months in patients receiving EDG-5506 10 mg once per day versus placebo. Secondary endpoints include timed function tests, change in fat fraction assessed by MRI, muscle damage biomarkers, and patient-reported outcomes.15
North Star Ambulatory Assessment: What Constitutes a Meaningful Difference?
Byrne described dystrophinopathies as a spectrum that includes both Duchenne and Becker. “Decreasing levels of dystrophin lead to significant impairment, and, ultimately, loss of ambulatory and cardiopulmonary function,”16 he said, adding that there was significant unmet need across the lifespan of both conditions. Explaining how the reductions in NSAA scores over time translated into an impact on everyday activities, he said losing the ability to run, hop, and jump stopped patients from playing sports, while the inability to stand on their heels hindered attempts to walk on uneven ground or cycle, for example. If someone cannot stand on one leg, they can have difficulty dressing themselves, and if they cannot stand from a chair, they can have difficulty using a toilet independently, getting out of bed themselves, or using public transport.
Data from baseline assessments of ARCH and CANYON participants show how patients with Becker move through NSAA stages, first developing compensation strategies before losing function in 15 of the score’s domains (unpublished data, presented at WMS 2023).12 “When we look at this, based on the functions that are lost, and where compensation comes into play, it gives us a better idea of how progression occurs over the years,” said Byrne.
The 39 study participants were segregated into those with a NSAA of ≥30 (Group 1), between 20–29 (Group 2), between 10–19 (Group 3), and those with the most impairment, or NSAA <10 (Group 4) (unpublished data, presented at WMS 2023).12 Once disease decline has started, natural history data have suggested that patients can expect to move from the midpoint of Group 1 to the midpoint of Group 2 over the course of around 5 years; from the midpoint of Group 2 to that of Group 3 over around 8 years; and from the midpoint of Group 3 to the midpoint of Group 4 over around another 8 years, Byrne explained.7,9
In Group 1, patients could complete all 17 assessed functions, though a small proportion (approximately 10%) had lost the ability to stand on their heels.12 There was also some weakness-related compensation in domains such as standing up from a chair, rising from the floor, and hopping on the right leg (unpublished data, presented at WMS 2023).12
Those in the NSAA 20–29 group could complete most functions, though compensation was recorded in many domains, reflecting the progressive loss of muscle and progressive weakness.12 “The compensation is particularly evident in standing up from a chair, which is one of the telltale early signs that bring patients to medical attention,” Byrne said. More than 80% showed compensation in either leg when descending or ascending a box step, 80% had difficulty with walking and running, and 100% had compensation for rising from the floor.12 At NSAA 10–19, which reflects the baseline function of those involved in the ARCH study, patients were unable to complete most functions, and needed to compensate for almost all functions (Figure 2).12 “This really reflects the progressive loss and progressive weakness as the disease continues and as individuals get older. At this point, 40% exhibit compensation during standing, all are having difficulty with standing from a chair, and some have lost that ability altogether,” said Byrne. 12 Patients in Group 4, who had a recorded NSAA of 0–9 points at baseline, had lost function in most domains. This cohort showed minimal ability to complete the activities, and was nearly non-ambulatory (unpublished data, presented at WMS 2023).12
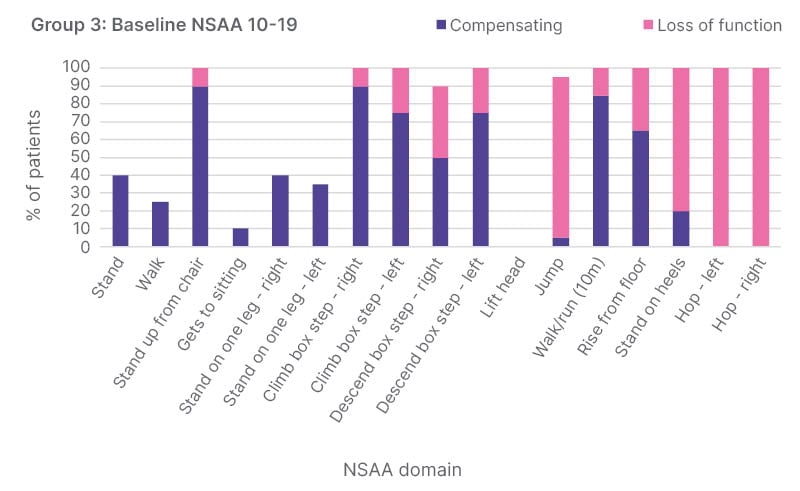
Figure 2: Loss of function and compensation at baseline in patients with North Star Ambulatory Assessment 11–20 (Group 3) enrolled in ARCH.
NSAA: North Star Ambulatory Assessment.
All the NSAA domains have a direct impact on a person’s ability to live their life well, Byrne said. “These activities obviously have real world implications for Becker patients, and their ability to participate in social activities, sports, and so on.”
As such, stabilising function and reducing the rate of decline is “an important life goal” for people living with Becker. Being able to provide this would help to address a significant unmet medical need in this condition, he concluded.






