Abstract
Senescence occurs as a part of the cellular response to different stressors. With increasing age, continuous exposure to stressors leads to age-induced senescence. Pancreatic β-cell proliferation and glucose homeostasis also decrease with age, which results in a decrease in β cell mass and, eventually, the possible development of diabetes. This process is mediated through impaired cell cycle regulators, along with specific increases in cell cycle inhibitors, telomere shortening, and defective DNA repair mechanisms. Diabetes contributes to β-cell senescence through hyperglycaemia, dyslipidaemia, oxidative stress, and inflammation. β cells isolated from patients with Type 2 diabetes mellitus have been shown to have senescence markers, such as senescence-associated secretory phenotype genes and β-galactosidase. In this paper, the authors discuss the mechanisms of cellular senescence, how senescence is impacted by the diabetic microenvironment, and the possible mechanisms and factors contributing to β-cell senescence.
INTRODUCTION
Cellular senescence is a stress response to remove cell damage through immune system activation. The term ‘senescence’ was originally coined in 1961 by Hayflick and Moorhead.1 Insufficient insulin production and secretion are key features of diabetes. Normal ageing leads to a decline in the replication of insulin-producing β cells in the pancreatic islets.2 However, a dysfunctional response affecting these β cells has been shown to be involved in diabetes pathogenesis. Decreased insulin secretion, owing to pancreatic cell dysfunction, has been linked to numerous factors, such as increased oxidative stress, endoplasmic reticulum stress, autoimmunity, and inflammation.2 Given that these are common characteristics of cellular senescence, it has been suggested that pancreatic β-cell dysfunction occurs through the induction of cellular senescence.3 In this review, the authors provide evidence for the involvement of cellular senescence in diabetes pathogenesis, its underlying molecular mechanisms, and the relationship between the diabetic microenvironment and β-cell senescence.
THE MECHANISM OF CELLULAR SENESCENCE
In aged tissue, altered gene expression promotes an increase in the secretion of proteins relevant to senescence. These proteins include senescence-associated secretory phenotype (SASP) proteins and senescence messaging secretome (SMS) proteins, such as amphiregulin, EGF, BMP, FGF, VEGF, and WNT. Csf1, IL8, Cxcl15, and Ccl2 proteins activate immune cells, including natural killer cells and macrophages. Moreover, the secreted proteins upregulate proteases (MMP, elastin, plasminogen activators, and collagen), survival factors, immune modulators, chemokines, and cytokines (for example, CXCL1, IL-6, IL-1β, IL-8, CXCL2, IL-1α, and CCR2 receptors).4 Lastly, SASP proteins shed cell surface molecules, inflammatory growth factors, and exosomes in the tissues’ microenvironment, increasing the overall stress response.4 As DNA damage causes senescence through reactive oxygen species or oncogenic signalling,1 oncogenic activation of RAS protein results in DNA damage, telomere shortening, and oxidative stress. DNA damage activates the DNA damage response, inducing p21Waf1/Cip1 and p16Ink41 cyclin-dependent kinase inhibitors, leading to cell cycle arrest (Table 1).5-9
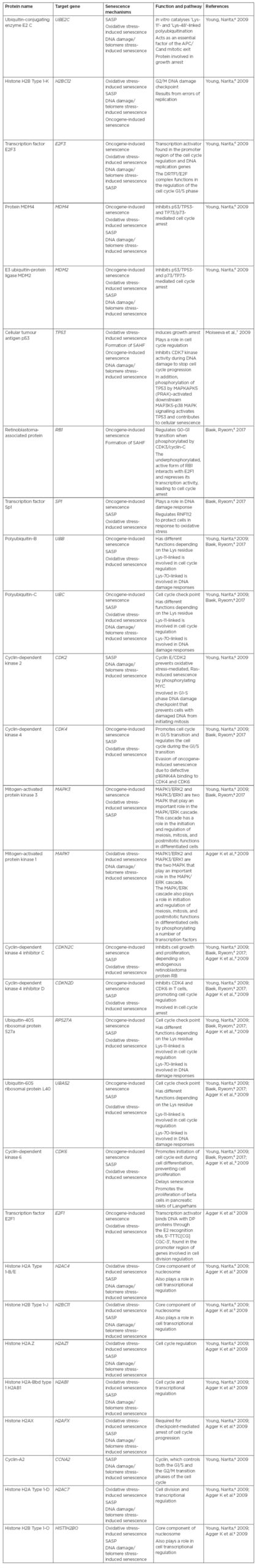
Table 1: Proteins involved in the stimulation of cell senescence signalling pathways.
APC/C: Anaphase promoting complex/cyclosome; SAHF: senescence-associated heterochromatin foci; SASP: senescence-associated secretory phenotype.
THE DIABETIC MICROENVIRONMENT MEDIATES CELLULAR SENESCENCE
The diabetic microenvironment is characterised by chronic hyperglycaemia; imbalanced lipid breakdown (dyslipidaemia); increased formation of reactive oxygen species;10 and a distinct, reduced antioxidant potency that leads to cellular inflammation. This state of simultaneous glucotoxicity, lipotoxicity, and inflammation paves the way for the micro- and macrovascular complications of diabetes.11
Aguayo-Mazzucato et al.12 isolated β cells from 8-month-old rats based on the activity of the senescence marker β-galactosidase (β-gal). β-gal-positive cells showed upregulation of senescence genes and a decline in β cell markers, such as Ins1, Mafa, Nkx6.1, and Pdx1. SASP genes, such as TNF and CXCL1, were upregulated in β-gal-positive cells. An increase in p16Ink4a was also observed in conditioned media from β-gal-positive cells. In β-gal-positive cells in mouse models of insulin resistance, SASP were acquired through either continuous administration of S961, an insulin receptor blocker, or through a high-fat diet. In the S961 model, there was a reversal of all the previous parameters, including a decrease in SASP gene expression back to the normal level within 2 weeks of terminating the drug. When the research group tested senolytic therapy by administering B/B homodimeriser, which causes a loss of cells expressing p16Ink4a, an improvement of β-cell senescence and a reduction of SASP parameters in aged mice were achieved. Glucose-mediated insulin secretion was also improved.12
β-gal positivity was higher in islet cells of older patients and patients with Type 2 diabetes mellitus. SASP, such as CCL4 and IL-6, beside p16Ink4a, were increased in β-gal-positive cells. The senescence marker insulin-like growth factor 1 receptor (IGF1R) was found to be higher in islets from patients with Type 2 diabetes mellitus <40 years of age; this denotes premature β-cell senescence. Additionally, the expression of p53-binding protein 1 (p53BP1) was higher in islets from patients with Type 2 diabetes mellitus.13 Aguayo-Mazzucato et al.12 reported a differential expression in age-related genes in young compared to older β cells; whereas Kcnq5 and Fgfr1 were upregulated in younger mice, while IGF1R, CD99, and Bambi were upregulated in older mice. An observed age-dependant increase in IGF1R staining in the human pancreata was also observed,13 and aged islets displayed better staining with IGF1R, which was associated with defective insulin secretion.13 This may indicate premature β-cell senescence in patients with Type 2 diabetes mellitus.13 These markers, IGF1R and p53BP1, showed heterogenic distribution in older β cells of the same islet and between islets.13
MECHANISM OF AGEING OF β CELLS
Age-Associated β-Cell Growth Arrest
The balance between β-cell proliferation and growth arrest is important in maintaining a proper β cell mass sufficient for glucose haemostasis. While β cells normally have a low replication rate, under certain conditions, such as pancreatic injury, their proliferation increases to cope with the pancreas’ deficient metabolic function.14 Physiologically and with increased age there is a decrease in β cell replication.15 Similarly, with age the human pancreata showed deteriorated β-cell replication15 and a decrease in the expression of cell cycle and cell proliferation regulator FoxM1.16 Also, with β-cell ageing, there is a decrease in the expression of pdx1, the transcription factor which is responsible for β-cell division and maturation.15 Isolated human pancreata from healthy donors aged 7–66 years showed a decline in β-cell proliferation starting at age 16 years.15 Also, pancreata at autopsy from 124 individuals with diabetes and obesity aged 61–83 years showed reduced β-cell replication.17 The relationship between the decrease in β cell mass and signs of diabetes is still under investigation. In healthy donors with impaired fasting glucose level, there was a decrease in β cell mass.17 In another study, there was a reduction in β cell mass by 24% in patients with diabetes after 5 years of disease onset, which became worse with time.18 This decrease in β cell mass may be inherent because of defects in cell cycle regulators such as CDKN2A and CDKN2B. These genes reduce β-cell proliferation as a compensatory mechanism during maturation.19 β-cell dysfunction associated with diabetes and defective insulin secretion leads to impaired glucose clearance, further causing a reduction in β cell mass.20
Mechanism of Age-Related β-Cell Growth Arrest: Cell Cycle Mechanism
Any insult to the genetic material caused by stress leads to suspension of the cell cycle. The passage of the cell cycle from G1 to S, then from S to G2, and then finally to mitosis, is regulated by cell cycle checkpoints. These checkpoints grant the cells the time needed to correct any damage to their DNA. This mechanism prevents the passage of damaged or mutated genetic materials to the next cell generations. A set of enzymes called cyclin-dependent kinases (CDK) control and regulate the cell cycle.21 CDK are protein kinases that are only active when complexed with their cyclin subunit and are inactive when alone.22
Mechanism of Age-Related β-Cell Growth Arrest: Cell Cycle Regulation in β Cells
β cell cycle activators
D-cyclins are cycle activators which regulate cell proliferation by regulating the cell’s progress from the G to the S phase.23 D-cyclins include D1–3 cyclins that complex with CDK4. Cyclin D2-null mice suffer from decreases in islet mass and, later in life, diabetes owing to a severe reduction in β-cell proliferation.23 In the absence of cyclin D2, cyclin D1 could partially replace cyclin D2. In the double mutant form, severe reductions occur in β-cell proliferation and diabetes follows early in life. Cyclin D is upregulated, as is β-cell proliferation, in murine β cells following partial pancreatectomy, which serves as a model of β-cell regeneration. Transgenic mice in which cyclin D1 or D2 were overexpressed showed enhanced β-cell proliferation.24,25 Significant increases in β-cell proliferation were observed in mice that expressed a single point mutation at site 280 of cyclin D2, promoting its consistent activation, regardless of age. Using adenovirus to overexpress cyclin D1 in human and rat islets significantly enhanced β-cell proliferation. In contrast to rodent cells, human β cells showed high expression of cyclin D3, with lower expression of cyclin D1 or D2.26 When overexpression of cyclin D3 in human islets in vitro was complexed with CDK6, but not other cyclins, a significant increase in β-cell proliferation was observed.26
Cyclin-CDK interactions led to a reduction in cell cycle inhibitors, thus inducing cell proliferation.27 The β cell mass of CDK4-null mice showed a severe reduction, both postnatally and at 17 weeks, reaching only 10% when compared to the β cell mass of the wild type.28CDK4-knockout mice showed smaller organs and pancreata, indicating replication defects;29 and developed diabetes because of their defective β-cell replication, causing a defective β cell mass. Interestingly, this effect was not due to apoptosis or autoimmunity.29 Transgenic mice with mutated CDK4, so as not to be inhibited by cell cycle inhibitor p16INK4a, showed up to a 10-fold increase in islet size. However, this increase was not associated with an increase in glucose-induced insulin secretion.30 Sertad1 (Sei1) is a protein that is expressed in pancreatic islets and aids cyclin/CDK4 stabilisation through the inhibition of p16INK4a.31 Sei1-null mice showed defective insulin production owing to a reduced islet cell mass, which was also found to be independent of apoptosis. Using adenovirus to deliver CDK4 into human or rat β cells resulted in enhanced β-cell proliferation, which further increased when accompanied by cyclin D1 transfection.32 Knock-in mice in which CDK4 was upregulated showed enhanced β-cell proliferation and β-cell hyperplasia.30 Using CDK4R24C, β-cell specific knock-in mice also showed the previous results.33 CDK4 knock-in also resulted in the reversal of diabetes in obese mouse models (db/db41) through the enhancement of β-cell replication.34 This replication was improved after partial pancreatectomy in CDK4R24C mice,35 indicating the therapeutic potential of CDK4.36
The CDK5R1 gene encodes p35, which is responsible for the activation of cdk5. In Type 2 diabetes mellitus, CDK5R1 becomes hypomethylated and consequently activated, leading to upregulation of CDK5. CDK5 activation causes defective expression of Pdx1 that subsequently leads to failed mature β-cell maintenance and may lead to a form of maturity-onset diabetes of the young (MODY4).36
CDK6 was not detected in murine islets but was shown to be critical for β-cell replication in humans.26 The effect of age on cyclin D and CDK levels has not yet been examined. CDK2 is expressed in pancreatic islets, but its level is downregulated with age.28 In mice, CDK2 level decreased significantly at 11 months of age in comparison to a younger age.37 Deletion of CDK2 resulted in enhanced islet senescence and, eventually, diabetes.38
β cell cycle inhibitors and β-cell senescence
Cell cycle inhibitors include the INK4/ARF family: p16INK4a, p15INK4b, p18INK4c, and p19INK4d; as well as the CIP/KIP family: p21Cip1, p27Kip1, and p57Kip2. The INK4/ARF family interrupts D cyclin binding to CDK4 and CDK6, while the CIP/KIP family acts by binding to cyclin D, E, and A, and CDK.39 Overexpression of murine CDK inhibitors (CDKI), through induced overexpression or inhibition of their epigenetic regulators, resulted in cell cycle arrest and β-cell ageing.40 On the other hand, loss of CDKI or their epigenetic regulators led to islet hyperplasia.41,42
β cell cycle inhibitors and β-cell senescence: p27Kip1
Postnatally, the level of p27 was found to be upregulated in mature β cells. Postnatal deletion of p27 resulted in a significant increase in β-cell proliferation and a consequent increase in islet mass;43 no increase in islet mass or hyperplasia was observed in adult mice, indicating a state of postnatal β-cell quiescence, mediated by cell cycle arrest. After induction of diabetes using streptozotocin, p27-null mice showed increased β-cell proliferation and reversal of diabetes.42 In two other diabetes models, accumulation of p27 in the nucleus was noted in β cells.41 Additionally, p27-deficient mice exhibited enhanced glucose tolerance that was further improved by the deletion of p27.44
In this model, the deletion of p27 allowed for β-cell replication and prevented the development of diabetes.44 Using the mouse model of overexpressed p27, specifically RIP-CDKn1b in β cells, a significant decrease in cell replication was shown, resulting in the development of diabetes.41 Furthermore, cadaveric islet samples from individuals with diabetes showed increased nuclear p27, indicating that p27 is a main regulator in response to increased β-cell requirement.41
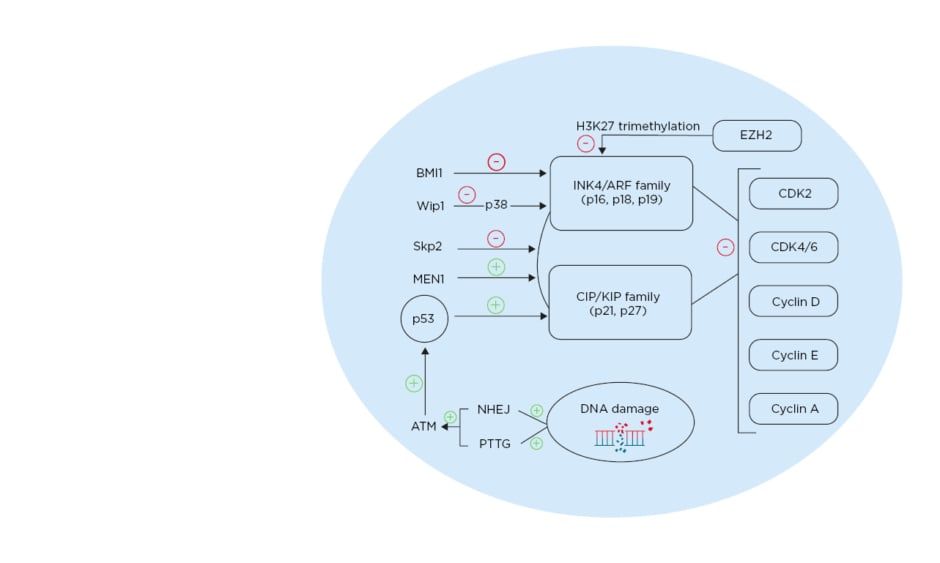
Regulation of p27 is critical in the cell cycle; it is regulated by E3 ubiquitin ligase S-phase kinase-associated protein 2 (SKP2) and is degraded through E3 ubiquitin ligase SKP2 ubiquitination. In β cells lacking SKP2, p27 upregulation is concomitant with a decrease β-cell proliferation and with senescence, leading to the development of diabetes.45 The multiple endocrine neoplasia type 1 (MEN1) tumour suppressor gene acts through a complex with histone methyltransferase to enhance expression of certain genes.46MEN1-null mice (MEN1-/-) showed increased β-cell proliferation and islet hyperplasia.47 Individuals with mutations in Menin, the product of MEN1, present with pancreatic islet tumours. Mice that are heterozygous to MEN1 (MEN1+/-) showed simultaneous decreases in the level of p27 and p18, resulting in islet hyperplasia (Figure 1).46,48
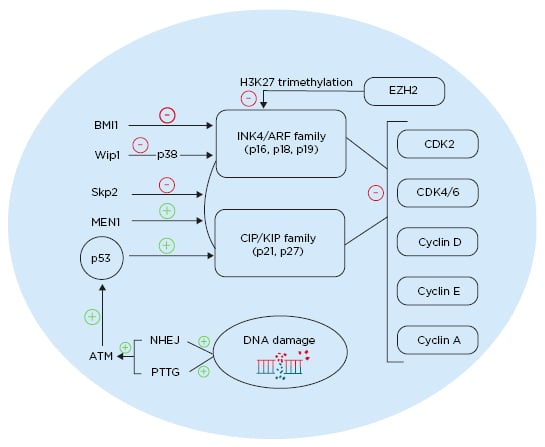
Figure 1: Different pathways affecting cell cycle regulation in pancreatic β cells. ATM: ataxia telangiectasia mutated gene; BMI1: BMI1 proto-oncogene; CDK: cyclin-dependent kinase; EZH2: enhancer of zeste 2 polycomb repressive complex 2 subunit; MEN1: multiple endocrine neoplasia tumour suppressor gene; NHEJ: non-homologous end joining; PTTG: pituitary tumour transforming gene; Skp2: S-phase kinase-associated protein 2; Wip1: wild-type p53-induced phosphatase 1. Created with BioRender.com.48
β cell cycle inhibitors and β-cell senescence: p21
p21 is a CDKI which halts the cell entrance to the S phase.49 p21 is the downstream effector of the p53 senescence pathway. Upregulation of CDK4 or cyclin D1 increased p21 levels in the nucleus, denoting its importance in regulating the cell cycle.50 However, in a murine model of p21 deficiency, normal islet proliferation took place, indicating that there may be other cell cycle regulators that compensate for p21 function.51 Minamino et al.52 showed that oxidative stress enhances the expression of p21.
Treatment of isolated rat β cells with hydrogen peroxide led to elevated levels of p21 and suppression of insulin production.53 Induction of p21 in rat islets was followed with induction of diabetes.53
β cell cycle inhibitors and β-cell senescence: p16
With age, β cells show upregulation of cell cycle inhibitors, such as the tumour suppressor protein p16Ink4a.54 CDK4 and CDK6 complex with cyclin D, phosphorylating pRB and activating the transcription factor E2F, which permits the transition from G to S phase. With age, p16Ink4a sequesters CDK4 and CDK6 and thus inhibits interaction with D cyclins.54 Transgenic mice with β cells overexpressing p16Ink4a showed significant reduction in β-cell replication at 26–32 weeks, comparable to older mice.54 p16Ink4a-null mice showed no age-related reduction in β-cell replication; β cells from mice of 60 weeks of age proliferated similar to younger mice that were 10 weeks of age.54
p16 gene locus INK/ARF is epigenetically regulated by polycomb-group proteins. These proteins regulate stem cell self-renewal by modifying the INK/ARF locus.55 Expression of BMI-1 polycomb proteins decrease with age and have an inhibitory role on p16. Knockout of BMI-1 resulted in higher expression of p16 and p19, along with a consequent decrease in β-cell proliferation. Furthermore, inhibition of BMI-1, caused by premature senescence due to an increased expression of p16, led to a decrease in β cell mass and impaired glucose tolerance (Figure 1).48,56
The enhancer of zeste homology 2 (EZH2) is another polycomb protein that is more expressed in younger mice. It acts as an inhibitor of p16 and p19 through H3K27 trimethylation of Ink4a and Arf.40EZH2-null mice show a decrease in β-cell proliferation and eventually develop diabetes.40EZH2 and p16 double-null mice were able to reverse this diabetes.
p38 mitogen-activated protein kinase (p38 MAPK) is expressed in response to cellular stressors.57 p38 MAPK inhibits BMI-1 and thus p38 activation, leading to an increase of p16 and p19.58 Aged islets showed more upregulated p38 MAPK compared to younger islets.59 In p38 MAPK-deficient mice, global inhibition of CDKI was observed with age.59 Induction of diabetes using streptozotocin in these mice showed partial reversal, caused by enhanced β-cell proliferation. The oncoprotein Wip1 is an inhibitor for p38 MAPK and its level is reduced with ageing. Thus, deletion of Wip1 in mice resulted in an upregulation of p38 MAPK and development of ageing-associated diabetes. However, induced expression of Wip1 resulted in a decrease in p38 MAPK and, consequently, a decrease in p16 expression, p19 expression, and the reversal of diabetes (Figure 1).48,59
CELLULAR STRESSORS AND β-CELL SENESCENCE
The Role of Telomere Shortening
Telomeres are repeated DNA sequences located at the end of chromosomes that shorten with every cell cycle as a result of double-stranded breaks and DNA damage. These breaks activate the p53 apoptosis pathway or p21-mediated senescence.60 In mice deficient in the telomerase RNA template (TERC), an impaired glucose tolerance occurred that was partially corrected by deleting p53.61 In these mice, β-cell proliferation and islet mass decreased, which were caused by telomere shortening due to deficient TERC.62TERC-null mice who were exposed to β cell stress through a high-fat diet showed significantly impaired glucose tolerance associated with insulin insensitivity.52 Higher levels of p53 and cellular ageing in the fat pads of these mice may have contributed to the observed insulin resistance.
DNA Repair Dysfunction
DNA repair mechanisms, such as nonhomologous end joining (NHEJ), are the main pathway for repairing double-strand breaks.63 Deletion of DNA ligase IV, which is responsible for fixing the DNA ends as a part of the NHEJ pathway, leads to the activation of p53, p21-mediated β-cell senescence, decreased proliferation, and, eventually, diabetes.64 Individuals with diabetes thus show higher DNA damage, concomitant with reduced DNA repair efficiency.65
The NHEJ pathway effectors can be subdivided into two groups: the DNA end recognition and processing complex, composed of the DNA-dependent kinase catalytic subunit (Ku70/80 heterodimer and DNA-PKcs) and the DNA repair factor Artemis; and the DNA ligation complex (DNA ligase IV, XRCC4, Cernunnos/XLF).63 Ku70/80 have the ability to bind and phosphorylate the pancreatic duodenal homeobox-1 protein (PDX-1), which is the main transcription factor in pancreatic development, through recruitment of DNA-PKcs.66 An increase in DNA damage stimuli leads to increased DNA-PKcs and PDX-1 degradation. Defective NHEJ pathways were reported in deficient pituitary tumour transforming genes.67 In mice deficient in pituitary tumour transforming genes, upregulated p53 and p21 levels concomitant with increased DNA damage were observed, and this led to β-cell senescence.68
Ataxia telangiectasia mutated protein (ATM) is a protein kinase and cell cycle regulator that acts upon DNA damage. It phosphorylates and activates many effectors, including tumour suppressor p53, and thus aids in DNA repair and prevention of cellular senescence.69 In ATM-deficient mice, impaired glucose tolerance and, eventually, diabetes developed.70 Induction of ATM in β cells without DNA damage using chloroquine injection in two diabetes models (ob/ob, db/db) reduced fasting and random blood glucose.71 Oxidative stress also enhanced ATM regulation of metabolic activity, and thus prevented development of diabetes induced by oxidative damage of β cells.72
CONCLUSION
In this review, the authors provided an overview of how the diabetic microenvironment causes cellular senescence and the relation of β-cell senescence to the development of diabetes. CDKI upregulation may induce β-cell senescence; thus, inhibition of CDKI may represent a therapeutic approach to enhance β cell mass. MEN1 deletion increased β-cell proliferation through inhibition of the CDKI p27 and p18. In addition, gestation-induced β-cell proliferation was caused by decreases in MEN1, reflecting a possible therapeutic effect of MEN1 in diabetes. Regulating Wip1 impacts ageing-associated β-cell proliferation, as induction of Wip1 resulted in a decrease of p38 MAPK. Enhancement of Wip1 may thus present a promising therapeutic approach for diabetes. The pathogenesis of patients with Type 2 diabetes mellitus and β-cell senescence was related to an increase in SASP factors. However, more studies are needed to directly relate the connection between SASP factors with the pathogenesis of diabetes in aged β cells, and the role of senolytic therapies in the reversal of impaired β-cell proliferation in ageing individuals.