INTRODUCTION
Kikuchi–Fujimoto disease (KFD), histiocytic necrotising lymphadenitis, is a benign and self-limiting disease characterised by lymphadenopathy, fever, fatigue, and leukopenia. KFD typically affects young Asian females. The symptoms of KFD can also mimic more sinister conditions such as non-Hodgkin lymphoma, tuberculous lymphadenitis, or reactive lymphadenopathy.1,2,3 The pathogenesis of KFD is unknown, but there are viral or autoimmune aetiologies. Several reports have emphasised the importance of KFD and systemic lupus erythematosus (SLE) associations.1,4
CASE REPORT
A 26-year-old Caucasian male was admitted to the outpatient clinic with erythematous and infiltrative lesions on the right cheek and forehead, as well as in the left retro auricular area, with an irregular shape, peeling on the periphery, without subjective symptoms for 6 months.
Nine years ago the patient was treated with lymecycline and isotretinoin due to cystic acne with improvement, which initially improved. However, the condition worsened again 6 years ago, and the patient was treated with isotretinoin (0.5 mg/kg body weight).
Eight years earlier, he was diagnosed with lymphadenopathy of the cervical and submandibular nodes (up to 2 cm) with leukopenia 2.21 G/l, elevated transaminases (alanine transaminase exceeded the upper limit of normal by 6.6 times, aspartate aminotransferase by 4.3 times), moderately elevated lactate dehydrogenase values, and antibodies against Epstein-Barr virus in the IgG class. Based on histopathological examination of the lymph node, trepanobiopsy, and additional tests excluding the lymphoproliferative process, KFD disease was diagnosed as histiocytic, necrotising lymphadenitis, and the liver damage was considered to be the result of Epstein-Barr virus infection. The symptoms of KFD lasted for about 1.5 years and resolved spontaneously.
Laboratory tests showed no significant deviations from the antinuclear antibody (1:160 normal), and the immunoblot was negative. The histopathological examination of the lesion on the forehead revealed features occurring during lupus erythematosus but the patient did not report any other symptoms typical of SLE. A diagnosis of cutaneous chronic lupus erythematosus was made, and the patient received 200 mg hydroxychloroquine once a day and methylprednisolone acetate topically, and showed good tolerance and improvement within a month. The patient was advised to avoid UV exposure and to undergo regular check-ups every 6 months (Figure 1).
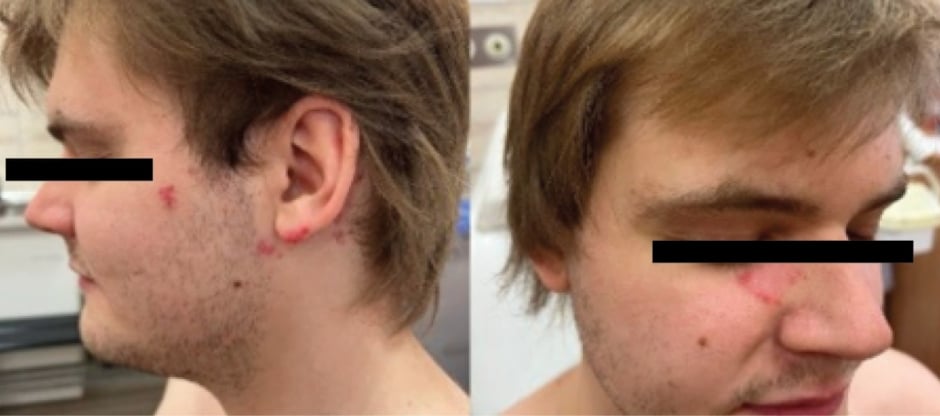
Figure 1: Chronic cutaneous lupus erythematosus in a patient with a history of Kikuchi–Fujimoto disease.
DISCUSSION
KFD can occur in patients with preexisting SLE, or it can coexist with SLE, or evolve into SLE. In some cases, histologic and immunohistochemical features of SLE lymphadenopathy are indistinguishable from KFD. Therefore, antinuclear antibody screening is recommended at diagnosis and close follow-up, especially in patients with cutaneous lesions, for the early detection of an autoimmune disease.1-4
The authors’ patient had skin lesions typical of chronic lupus, without general symptoms, and the patient didn’t fulfil SLE diagnosis criteria according to both the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) classification. However, early initiation of hydroxychloroquine may prevent the development of SLE in the future. Unfortunately, there is no current literature advising how best to monitor patients with KFD for long-term complications and disease associations, such as SLE.
CONCLUSION
The authors present this case to highlight the rare clinical entity of KFD in the Caucasian population, the need for a long-term follow-up, and the high risk of associated autoimmune diseases, such as SLE. The authors’ patient only developed symptoms of chronic lupus approximately 7 years after the symptoms of KFD disappeared. Therefore, long-term follow-up of patients with this syndrome seems necessary.





