Abstract
Background
Left ventricular myocardial crypts are defined as invaginations traversing >50% of the myocardial wall, with an incidence of 9.1% in the general population. Several studies have suggested that these morphological findings could be a marker of myocardial disease. The authors present two cases of cardiac arrest (CA) due to ventricular fibrillation (VF) and incidental findings on cardiac magnetic resonance (CMR) of myocardial crypts, discuss the diagnosis, the possible correlation with hypertrophic cardiomyopathy, and hypothesise the significance of this entity.
Case Presentation
Case 1 discusses a 58-year-old male affected by arterial hypertension, with a family history of hypertrophic cardiomyopathy (HCM), who presented to the emergency department for a CA due to VF. The 12-lead electrocardiogram was negative for ST-elevation myocardial infarction, Bragada’s pattern, or QT time elongation. Blood gas analysis showed no ionic abnormalities and no hypoxia. Coronary arteries were free from stenosing atheromatous lesions at coronary angiography. CMR showed isolated left ventricular crypt with mild hypertrophy of myocardial wall, preserved ejection fraction, no hyperintensity in the short tau inversion recovery sequences compatible with oedema, and no sign of fibro-adipose tissue substitution at postcontrast sequences. An implantable defibrillator for secondary prevention was implanted and genetic testing was carried out for HCM.
Case 2 discusses a 59-year-old Peruvian male, with a family history of cardiovascular diseases, who suffered from CA due to VF while under elective ear surgical intervention. A defibrillation with a single DC shock was sufficient to restore sinus rhythm. The 12 lead electrocardiogram was negative for ST-elevation myocardial infarction, Bragada’s pattern, or QT time elongation. Transthoracic echocardiography showed mild ventricular hypertrophy and normal global and regional biventricular kinetics (60%), along with mild mitral regurgitation. Coronary arteries were free from stenosing atheromatous lesions at coronary angiography. At CMR, an isolated left ventricular crypt in the inferior basal wall with an extension of 7 mm LL was detected; there wasn’t hyperintensity in the short tau inversion recovery sequences compatible with oedema and no sign of fibro-adipose tissue substitution at postcontrast sequences was seen. The authors performed genetics testing for HCM, still ongoing. An implantable defibrillator for secondary prevention was implanted.
Discussion
These cases suggest that the presence of crypt could be a marker of myocardial disease. This should warrant clinical follow-up prior to the potential development of left ventricular hypertrophy and the occurrence of serious cardiovascular events, such as heart failure or death. With the evolution of new imaging techniques such as CMR and CT, the detection of these myocardial defects is more accurate. Genetic testing is useful to identify carriers of pathological mutation of HCM that need close follow-up.
Key Points
1. Myocardial crypts, defined as invaginations of the myocardial wall, are often considered markers of myocardial disease. In particular, they are associated with hypertrophic cardiomyopathy.
2. The authors present a case series of two patients who underwent resuscitated cardiac arrest without any explainable cause, and in whom myocardial crypts were found during heart MRI.
3. Myocardial crypts are frequent findings during autopsy or imaging studies but there is a lack of understanding of their prognostic significance in terms of cardiovascular risk or myocardial pathology. Future studies and observations using cardiac MRI will shed light on this topic.
INTRODUCTION
Left ventricular myocardial crypts are rare structural abnormalities and are defined as invaginations traversing major than a half of the myocardial wall, and are generally situated at the basal or mid-inferior portion of the septum or at the apical and in the medium segments of the ventricular wall.1-3 In a large study on the general population undergoing cardiac computed tomography, the incidence of crypts was 9.1%, comparable with previous studies on CMR investigation. This shows that crypts are not associated with major adverse cardiovascular events in an intermediate-term period of observation.4-5 Due to the increasing use of advanced imaging, they were found to be more frequently incidental findings than previously thought.5 It’s important to discriminate left ventricular (LV) crypts from similar isolated non-compaction cardiomyopathy, and myocardial diverticula. While crypts are present predominantly at the basal or mid-inferior portion of the septum and penetrate the myocardium perpendicular to the endocardial border, non-compaction is predominantly seen in the apex and mid-ventricular wall portion of LV and it’s a non-compacted stratum of myocardial muscle oriented parallel to another layer of compact myocardium.6-7
On the other hand, ventricular diverticula are described as congenital abnormalities, characterised by an extroflection of the myocardial muscle across the entire wall. Crypts and diverticula contract themselves simultaneously with systole, but the diverticula have a narrow attachment collar and extend widely beyond the walls of the ventricle and the external myocardial margin.6-8 The genesis or the process of the remodelling of myocardial crypts during life is unclear as there aren’t studies of imaging that document the evolution of these myocardial anomalies from childhood to adult life.
Their formation probably takes place during embryological modification of the myocardium, related to a failure of reabsorption of the trabeculated area of the myocardial ventricular wall.9 This is probably due to altered contractility properties of embryonic myocytes, which cause an unusual mechanical tension towards the insertion points, pulling apart the myocardial fibres and generating the crypts.10 Near crypts are usually seen in myocyte disarray, supporting the concept of disruption of normal myocyte disposition as a morphological substrate for crypt genesis.11
While CMR imaging is the gold standard diagnostic technique to detect myocardial abnormalities, transthoracic echocardiography (TE) can be an initial diagnostic instrument that may reveal some abnormalities or suspects that can guide further investigation.12-13 CMR imaging is better than standard 2D echocardiography in evaluating myocardial wall hypertrophy. Above all, in LV apical and anterolateral portions, it allows for a view of myocardial crypts and papillary muscle abnormalities, subclinical abnormal clues in patients with myocardial myofibrils gene mutations.14-16 Some studies highlighted that atypical presentation of HCM is more frequent than what was known.17-19 A particular entity is represented by focal segmental hypertrophy, a variant that sometimes involves only one or two myocardial segments, often with a non-contiguous pattern. In patients with HCM, it is noted that focal segmental LV hypertrophy is limited to the anterolateral free wall, posterior septum, or apex in 12% of cases.17-19
A lot of studies tried to show the correlation between findings of myocardial crypts or cleft and HCM, hypothesising that they could be a pre-phenotypic sign of that disease.20-22 A better understanding of these findings is essential. This has also been mentioned in the 2014 European Society of Cardiology (ESC) guidelines of hypertrophy cardiomyopathy, cited as a finding to pay attention to in patients who are suspected to have cardiomyopathy.14
Related to these recommendations the authors present two cases of patients who had a cardiac arrest (CA) due to ventricular fibrillation (VF), resuscitated promptly with direct current (DC) shock.
In both cases, the authors followed the current evidence and guidelines on the management of CA to identify the underlying aetiology, and to choose the best therapeutic strategy for the patient.23
CASE PRESENTATION
Patient 1
A 58-year-old Italian male affected by arterial hypertension, with a family history of HCM and without any previous cardiovascular history or significant comorbidities, presented to the emergency department after CA while doing sport. A single defibrillation with DC shock on VF was performed outside the hospital. On admission, a 12-lead electrocardiogram (ECG) was negative for ST elevation myocardial infarction (STEMI), any Bragada’s pattern, or QT time elongation.24-25 The ECG tracing was not suggestive of HCM or arrhythmogenic dysplasia of the right ventricle or LV hypertrophy.26
Blood gas analysis showed no ionic abnormalities (K+ 3.8 mmol/L with a normal value of 3.6–5.2 mmol/L) or signs of hypoxia. Coronary angiography showed coronary arteries free from stenosing atheromatous lesions.
During hospitalisation, the patient was subjected to a flecainide test to exclude a possible implication of underlying Bragada’s syndrome, and the results for Bragada’s ECG pattern were negative.27 Moreover, the authors performed TE with contrast (sulphur hexafluoride) to obtain the best definition of the LV kinetics, with a supplementary analysis with speckle tracking, which showed mild hypertrophy (maximal diameter of 12 mm at basal septum), normal biventricular kinesis (ejection fraction 60%) and volumes, and excluded abnormalities of valvular apparatus. An isolated distal anterior myocardial crypt was found.
CMR was performed because of the last finding and an isolated LV crypt in the mid-apical site of the septum with an extension of 9×6 mm antero-posterior (AP) x latero-lateral (LL) was detected (Figure 1); at that level, the ventricular wall was focally thinned. The papillary muscles appeared apicalised and bifid. STIR sequences didn’t highlight any compatible feature with oedema and no sign of fibro-adipose tissue substitution at postcontrast sequences was seen. During the observation, in the authors’ department at the ECG telemetry, no ventricular or supraventricular arrhythmias were detected, so the administration of anti-arrhythmic drugs was not required.
After the exclusion of the primary cause of CA, and considering the clinical suspicion based on instrumental findings, the patient underwent implantable cardioverter defibrillator (ICD) implantation for secondary prevention. Based on clinical suspicion of HCM, genetic testing was carried out for the patient.
Patient 2
A 59-year-old Peruvian male, with cardiovascular risk factors, a family history of cardiovascular diseases, and no previous cardiovascular events or significant comorbidities, suffered from CA due to VF while under elective ear surgical intervention and was treated by single defibrillation with DC shock, restoring sinus rhythm.
The 12-lead ECG excluded STEMI, Bragada’s pattern, or QT time elongation.24,25 The ECG pattern was not suggestive of HCM or arrhythmogenic dysplasia of the right ventricle, and there were no signs of LV hypertrophy found in ECG.26
No other reversible causes were identified, and no ion abnormalities at blood gas analysis, or other laboratoriesblood alteration.
TE showed mild ventricular hypertrophy (maximal diameter of 12 mm at basal septum), normal global and regional biventricular kinetics (left ventricular ejection fraction 60%), along with mild mitral regurgitation but without valve prolapse.28
Coronary arteries were free from stenosing atheromatous lesions at coronary angiography and the Flecainide challenge test was negative for Bragada’sECG pattern.27
A CMR was performed to further characterise this finding, which showed an isolated LV crypt in the inferior basal wall with an extension of 7 mm LL (Figure 2); STIR sequences didn’t highlight any compatible feature with oedema or signs of fibro-adipose tissue substitution at postcontrast sequences. During the observation, in the authors’ department at the ECG telemetry, no ventricular or supraventricular arrhythmias were detected. The subsequent clinical course was regular.
Like the previous case, the patient was first implanted with an intravenous unicameral ICD for secondary prevention and genetic testing for HCM was carried out.
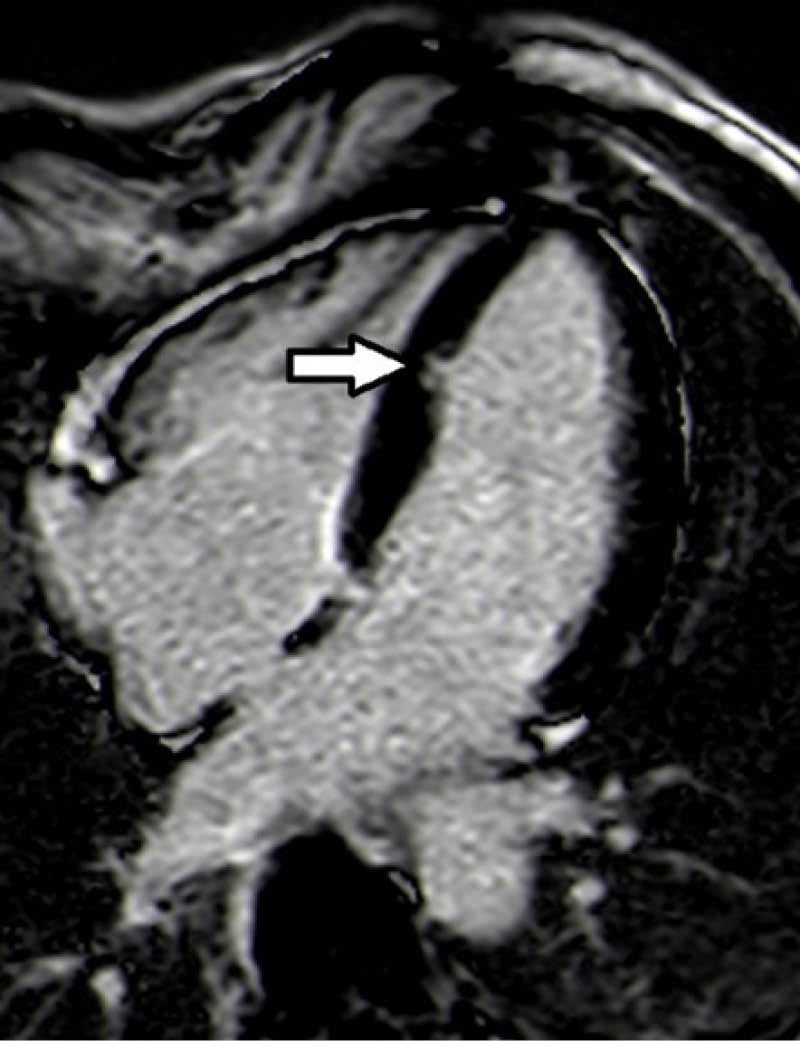
Figure 1: Four-chamber phase-sensitive inversion recovery sequence showed left ventricular crypt in the mid-apical site of the septum (dimension of 9×6 mm antero-posterior x latero-lateral).
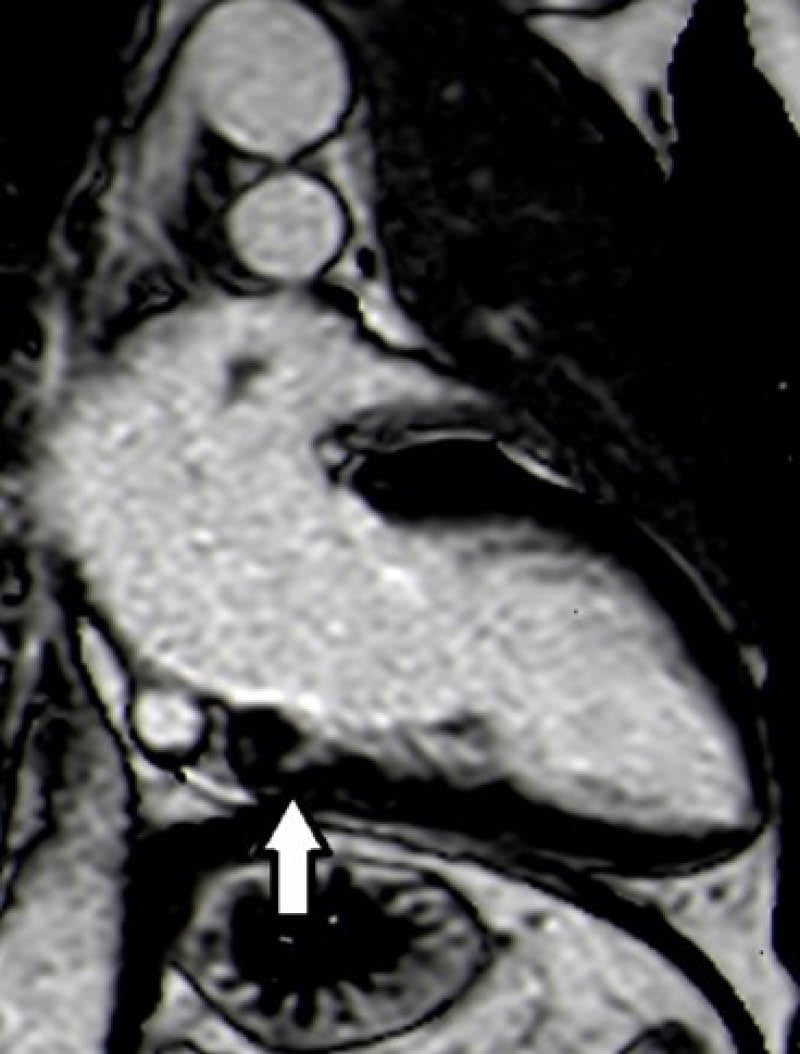
Figure 2: Two-chamber phase-sensitive inversion recovery sequence showed left ventricular crypt in the inferior basal wall (dimension of 7 mm latero-lateral).
DISCUSSION
HCM is the most frequent genetic cardiovascular disease.16 According to the ESC guidelines, HCM it is a pathological condition that occurs when myocardial thickness in any myocardial segment is ≥15 mm, with not explained by conditions of increased post-load. In case of minor wall thickness (13–14 mm), further evaluation including family history for syncope, sudden death, genetic tests, and ECG suggestive pattern is required.14 HCM exhibits the classic characteristics of a monogenic disorder that follows an autosomal dominant mode of inheritance. Changes in more than a dozen genes that code for proteins associated with the sarcomere are responsible for the development of HCM. The genes MYH7 and MYBPC3 are the most frequently occurring, accounting for around half of all patients with familial HCM. Meanwhile, TNNT2, TNNI3, and TPM1 mutations are relatively rare, contributing to less than 10% of HCM cases. Additionally, ACTC1, MYL2, MYL3, and CSRP3 mutations are also established causes of HCM, although they are uncommon. There is strong evidence for the role of these nine genes in HCM.
Other genes mutations have also been linked to HCM, but they are typically found in sporadic cases and small families.29 HCM is also characterised by multiple phenotypic and genotypic heterogeneity.17 The clinical presentation of HCM is variable and while several patients with HCM are asymptomatic, others may present even with sudden cardiac death (SCD) as their first clinical symptom, most commonly because of ventricular arrhythmias.8,16 In these cases, the authors present two males with CA, without any reversible or secondary identifiable cause like systemic inflammatory disease or toxic cause, with CMR finding of myocardial crypt; the first located in the mid-apical site of the septum, the last one in the inferior basal wall. Both cases didn’t show a frank hypertrophy phenotype according to the definition of HCM, but the clinical presentation along with mild hypertrophy of the basal septum (12 and 13 mm) and crypt finding led the authors to hypothesise a correlation between myocardial crypts and initial stage of HCM. In both these cases, global cardiac mass was within the normal range. Indeed, the correlation between LV mass and maximal wall thickness was not very strong, and in patients with asymmetric HCM, especially when only a small portion of the LV is affected, LV mass can still be within the normal range.14
Prior studies and cases have suggested that myocardial crypts may serve as precursor events or indicators of HCM in individuals who do not exhibit clear manifestations of the condition.30 In a limited investigation, the identification of two or more hidden chambers on CMR provided a 100% accurate indication in predicting the presence of HCM mutation carriers.31
In 2014 a study was conducted with MRI, where the relationships between the positive genotype for HCM mutations and the heart MRI images of phenotypically negative patients were investigated.21 In this paper, the author demonstrates that myocardial crypts and anterior mitral valve leaflet elongation are the two parameters that most strongly associate with the presence of sarcomere gene mutations. Observations also suggest that MYBPC3 mutation carriers have a two-fold prevalence of crypts and less LV systolic cavity reduction compared with the other combined mutations.21
It is widely understood that, among individuals with HCM, the most prevalent cause of death is SCD resulting from VF or ventricular tachycardia (VT). This unfortunate outcome often impacts young individuals who present frequently without symptoms.32 With these two case reports the authors suspected an association between the finding of crypts in an apparent healthy heart with CA.
The possible causes of ventricular arrhythmias in HCM include intraventricular conduction dispersion, which is caused by varying sizes of cardiomyocytes; the presence of fibrosis and disarray, which provide alternative conduction pathways that promote re-entry; and disruptions in intercalated discs that disrupt the smooth transmission of action potentials between cardiomyocytes, leading to arrhythmias.33-35 It’s difficult to demonstrate that these mechanisms could be present in a structural and functionally normal heart, only with a mutation of HCM present and not that of Frank’s disease. The infrequency of these discoveries in patients with a history of CA can be attributed to the low survival rate in these individuals.36 A previous study has reported evidence of myocardial invagination in the autopsy of a young man’s death of CA with asymmetrical ventricular hypertrophy.37-38
At present, there are no case reports describing a clear link in the literature between the finding of myocardial crypts and increased susceptibility to the onset of ventricular arrhythmias or SCD.
An active search for structural changes in imaging, collection of databases with this information, and ideally an anatomopathological finding in the future will be able to help in giving a more specific prognostic role to myocardial crypts.
Against these suspicions, in 2021 a major study on this topic was published that demonstrated that myocardial crypts are commonly found among the general population and do not show any connection with significant adverse cardiovascular events in the medium term.4 However, there is an error; the limitations of the study can’t rule out the current author’s doubts. The population of this study was composed completely of Danish people, but there are some studies that demonstrate differences in mortality and incidence between racial groups for HCM.39
Further pathological investigations are required on heart tissue with HCM and various underlying causes, as well as normal hearts, to deepen knowledge of this entity. The role of myocardial anomalies in the risk of experiencing fatal arrhythmic events will become clearer in the future through further research, enhanced survival rates for CA, and the easier availability of advanced imaging techniques such as CMR and cardiac CT. Another crucial point in most cases is the reduced availability of genetic testing, with extremely long times to obtain results. Improvement in this aspect could guarantee major information on the association between certain types of mutation and phenotypical patterns in vivo, or the risk of sudden death.40-42
Furthermore, it would be reasonable to conduct a multicentre prospective registry involving a substantial patient population who underwent CMR imaging, to monitor clinical outcomes continuously. This registry, like the EuroCMR registry, could offer valuable insights into the possible clinical significance of crypts, specifically concerning HCM.15 According to literature data, the occurrence of ventricular arrhythmias and syncope is thought to be very rare in pre-LVH mutation carriers, and current risk stratification strategies and guidance do not include these individuals.4
CONCLUSION
In conclusion, the authors’ case studies highlight important cardiovascular consequences that could be associated to left myocardial crypt, in the absence of other pathological finding at second level diagnostic tool. The occurrence of VF and VT increases the risk of SCD, especially when it happens in extra-hospital conditions, and when an external cardiac defibrillator isn’t available. Early identification of these patients could be essential to manage the early stages of HCM and prevent malignant arrhythmias or SCD. The implantation of ICD is highly effective in terminating VF and VT, indicating that these devices have a role in the primary and secondary prevention of malignant arrhythmias. Genetic testing and genetic counselling are useful to identify carriers of pathological mutation of HCM that need close clinical and instrumental follow-up in good clinical practice.45