Abstract
Homozygous familial hypercholesterolaemia is a rare and severe genetic disorder affecting one in 300,000 people. Due to the very high cholesterol levels since birth, compromise of the aortic valve and atherosclerotic cardiovascular disease can develop during the first decade of life. An early diagnosis and aggressive treatment before vascular damage occurs is critical for the evolution of the disorder. In this review, the authors discuss current lipid-lowering therapies for the management of patients with homozygous familial hypercholesterolaemia, with special focus on safety, efficacy, and potential cardiovascular benefits.
INTRODUCTION
Homozygous familial hypercholesterolaemia (HoFH) is a rare and aggressive disorder, resulting from the inheritance of mutations in the two alleles of genes regulating the low-density lipoprotein receptor (LDL-R) function. In >90% of cases, familial hypercholesterolaemia (FH) is caused by variants in the LDLR gene, and less frequently in APOB, PCSK9, or LDLRAP1 genes.1 Prevalence of HoFH is one in 300,000 people.1,2 The diagnosis is based on extremely high LDL-cholesterol (LDL-C) levels, usually >500 mg/dL. In addition, cutaneous and tendon xanthomas can be detected before the age of 10 years, and both parents should have high cholesterol levels suggesting heterozygous FH (HeFH).3 This population is characterised by high frequency and very early onset of cardiovascular disease (CVD).4 In children, cardiovascular (CV) events, including aortic and supra-aortic valve disease and coronary heart disease, are found in almost 50% of cases around age 11 years.5 Usually, if individuals with HoFH are not treated, they may not survive beyond the third decade of life.3,4,6
The major determinant of survival in this population is on-treatment total cholesterol (TC) levels, which are 6.2-times higher in cases with TC >15.1 mmol/L (585 mg/dL) than those with TC <8.1 mmol/L (314 mg/dL).4 On the other hand, there is evidence suggesting great phenotype heterogeneity depending, in part, on the molecular defect. LDL-C levels may overlap those observed in HeFH, and CVD may occur later in life in patients with defective mutations with certain LDL-R activity.7,8
This review summarises the advantages and disadvantages of available treatments approved for patients with HoFH. For this purpose, the Medline/PubMed databases were systematically searched for literature published between 1982 and May 2021 to focus on more relevant data applicable to the current scenario of the management of HoFH. No restriction on publication type was applied. Due to the rarity of the disorder, consensus statements, guidelines, randomised controlled trials, open-label trials, and case reports were analysed. The databases were searched with the terms “homozygous familial hypercholesterolemia,” “lipid-lowering therapies,” “statins,” “ezetimibe,” “PCSK9 inhibitors,” “lomitapide,” “evinacumab,” “mipomersen,” “LDL-apheresis,” and “liver transplantation,” related to efficacy and safety in this specific population. All literature in English were included. The literature revision was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines 2020.9
TREATMENT GOALS
The main objective of LDL-C reduction in treating HoFH is to delay or prevent the development of atherosclerotic coronary artery disease and the compromise of the aortic valve. As HoFH is a rare disorder, it can be assumed that goals proposed for HeFH in different national and international guidelines could be applicable to this condition.3, 10-12 The 2014 European statement recommended an LDL-C target of <2.5 mmol/L (<100 mg/dL) in adults, <1.8 mmol/L (<70 mg/dL) in adults with atherosclerotic cardiovascular disease (ASCVD) or diabetes, and <3.5 mmol/L (<135 mg/dL) in children.3 The same LDL-C goals were considered in the Spanish statement, but a 50% reduction in LDL-C was also added.12 This percent reduction in LDL-C is also recommended by the National Lipid Association (NLA)11 and National Institute for Health and Care Excellence (NICE) in the UK.10 However, there is general agreement that these targets are very difficult to achieve with standard lipid-lowering therapies (LLT) in most patients with homozygous FH.
CURRENTLY AVAILABLE LIPID-LOWERING TREATMENTS FOR PATIENTS WITH HOMOZYGOUS FAMILIAL HYPERCHOLESTEROLAEMIA
The mechanisms of action and efficacies of discussed approved LLT for HoFH are outlined in (Table 1).
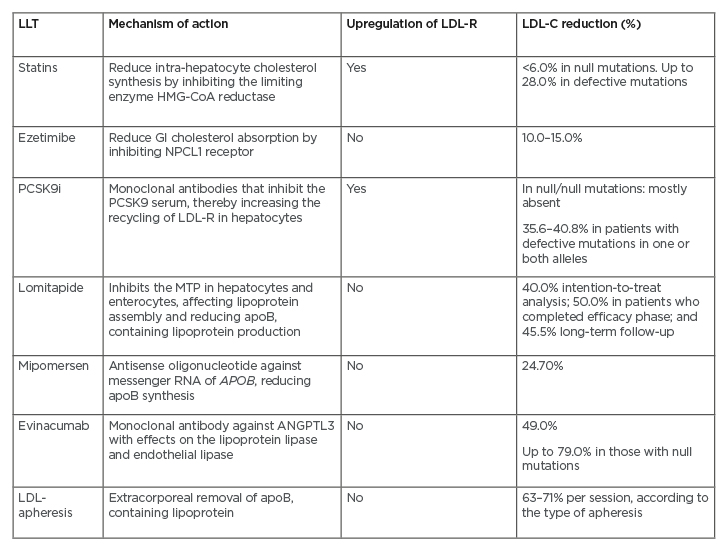
Table 1: The mechanisms of action and efficacies of approved lipid-lowering therapies for homozygous familial hypercholesterolaemia.
ApoB: apolipoprotein B; GI: gastrointestinal; HMG-CoA: 3-hydroxy-3-methylglutaryl-coenzyme A; LDL: low-density lipoprotein; LDL-C: low-density lipoprotein-cholesterol; LDL-R: low-density lipoprotein-receptor; LLT: lipid-lowering therapy; MTP: microsomal triglyceride transfer protein; NPCL1: Niemann-Pick C1-Like 1; PCSK9i: PCSK9 inhibitors.
Statins and Ezetimibe
Statins are the mainstay of treatment and should always be prescribed in patients with HoFH together with a healthy lifestyle, ideally starting at the age of 2 years. Statins upregulate LDL-R; therefore, the response is very modest in HoFH, depending on the severity of the causative mutation. LDL-C reduction with statins varies between 6% in those patients with null mutations and 28% in those carrying defective mutations.13 Randomised trials comparing efficacy and safety of statins in treating HoFH have included children aged from 6 years, and have shown that statins are well tolerated, with isolated cases of myalgia, no significant increases in transaminases, and no patient discontinuation of medication due to an adverse event (AE).13,14 Reduction in mortality has been shown retrospectively in a cohort of patients with HoFH in South Africa.6 Despite a mean reduction of 26% in LDL-C levels, mainly with statins, a 66% risk reduction in mortality and a significant delay in the age of first CV event or death were observed.6
Due to the severe hypercholesterolaemia, patients with HoFH require other LLT to further reduce LDL-C levels and reduce the residual cholesterol risk.
Ezetimibe targets the intestinal Niemann-Pick C1-Like 1 (NPC1L1) protein, selectively inhibiting cholesterol absorption without affecting the absorption of fat-soluble vitamins. It is commonly used in combination with statins, providing approximately 15–20% further reduction in LDL-C in the primary hypercholesterolaemia or high-risk population.15,16 There are few studies addressing the efficacy and safety of ezetimibe in treating individuals with HoFH aged 12 years and older.17,18 When ezetimibe is added to a high-intensity statin, with or without LDL-apheresis, there can be a further reduction of at least 20.5% in LDL-C levels.17 In general, the safety profile of ezetimibe co-administered with statins is similar to that observed with statins in monotherapy.
A recent systematic review including 26 randomised clinical trials showed that ezetimibe with statins reduced the risk of major adverse CV events by 6% compared with statins in monotherapy; however, the combination did not show benefit in reducing total mortality.19 In terms of safety, ezetimibe is well tolerated, and there are no differences in the risk of hepatopathy or myopathy and no risk of new-onset diabetes.20
PCSK9 Inhibitors
Alirocumab and evolocumab are monoclonal antibodies that lower plasma LDL-C levels by binding PCSK9 and upregulating LDL-R expression on hepatocytes. Several studies have shown that proprotein convertase subtilisin/kexin type 9 inhibitors (PCSK9i) reduce LDL-C levels in up to 60% of patients with HeFH and other high-risk populations.21-24 Moreover, two randomised controlled clinical trials in secondary prevention populations demonstrated that PCSK9i reduced ASCVD by 15% on top of a maximum-tolerated statin therapy compared with statins alone.21 Both agents were approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) to be used in patients with HeFH and in patients with ASCVD. Evolocumab was also approved for the treatment of patients with HoFH over age 12 years. The safety and efficacy of both PCSK9i in patients with HoFH were evaluated in Phase III trials25,26 and, in the long term, the open-label TAUSSIG study with evolocumab.27
In the TESLA trial,25 49 patients with HoFH aged >12 years and not on LDL-apheresis, were randomised to evolocumab 420 mg every 4 weeks (n=33) or placebo (n=16) for 12 weeks. A significant 30.9% LDL-C reduction was obtained with evolocumab compared with placebo. The response to evolocumab was related to the severity of the causal mutation. In those patients with one or two defective mutations, LDL-C reduction ranged from 40.8% to 46.9%. On the other hand, no response was observed in one patient with null/null mutations.
In the ODYSSEY HoFH study,26 69 patients were randomised to alirocumab 150 mg every 2 weeks (n=45) or placebo (n=24) for 12 weeks. A 26.9% reduction in LDL-C from baseline was observed with alirocumab at Week 12, and the difference with placebo was 35.6% (primary efficacy endpoint). In cases (n=5) with null/null mutations, the response was variable but mostly absent.
The TAUSSIG trial27 was an open-label, single-arm, non-randomised study that examined the long-term efficacy and safety of evolocumab in patients with HoFH and those with severe HeFH receiving stable LLT including LDL-apheresis (34 cases in HoFH). A 21% reduction in LDL-C levels from baseline was observed at Week 12 and remained constant until Week 216 (24% reduction). There was no difference in the response among patients with or without LDL-apheresis, and three HoFH cases on apheresis discontinued this procedure in the follow-up. Like in Phase III trials, patients with null/null mutations had variable responses that were lower compared to those with defective mutations.
In all trials, the safety and tolerability of PCSK9i were confirmed. No neutralising anti-PCSK9i antibodies have been detected with both evolocumab and alirocumab.
Lomitapide
Lomitapide has been approved by the FDA and the EMA for the treatment of adult patients with HoFH along with a low-fat diet and other LLT or LDL-apheresis. Lomitapide binds to and inhibits the microsomal triglyceride transfer protein (MTP), an enzyme with a key role in the assembly of very-low-density lipoprotein (VLDL) in hepatocytes and chylomicrons in enterocytes. By inhibiting MTP, lomitapide reduces plasma levels of VLDL, LDL, and chylomicrons.28 Therefore, the effect on lowering LDL-C levels is independent of LDL-R activity. On the other hand, its mechanism of action explains some AEs of lomitapide such as liver steatosis and steatorrhoea.29
Lipid-lowering efficacy and safety of lomitapide in treating HoFH has been demonstrated in Phase II and Phase III trials, in registries, and real-world experience reports.30-34 Patients receiving lomitapide are required to follow a very-low-fat diet (<20%). In the Phase II proof-of-concept trial,30 six patients without LLT received lomitapide at four different doses every 4 weeks. A dose-dependent reduction in LDL-C levels was observed, reaching 51% with the highest dose of 1 mg/kg. Also, triglycerides and apolipoprotein B (apoB) levels were significantly reduced by 65% and 55%, respectively. The most frequent AEs were gastrointestinal symptoms that were transient, dose-related, and explained mostly by low adherence to diet. An increase in transaminases was observed in four patients and fat accumulation in the liver was highly variable ranging from <10% to >30%. Transaminase and hepatic fat levels returned to baseline levels after the discontinuation of the drug in all patients. No patient withdrew from lomitapide due to an AE.30
The Phase III pivotal trial31 was a long-term, single-arm, open-label trial including 29 patients with HoFH >18 years old with molecular diagnosis. This trial had two phases: Phase I lasted 26 weeks to evaluate the efficacy; while Phase II lasted 52 weeks to evaluate long-term safety. In the efficacy phase, lomitapide was up-titrated every 4 weeks, from 5 mg to 60 mg every day or the maximum tolerated dose. Unlike the Phase II trial, all LLT including LDL-apheresis were maintained during the efficacy phase of this study and could be modified in the safety phase, while keeping the dose of lomitapide, reached in Phase I, stable. The median dose during the trial was 40 mg/day. Intention-to-treat analysis showed a mean 40% reduction in LDL-C levels, which rose to 50% in patients who completed the efficacy phase. LDL-apheresis did not affect efficacy. Four patients discontinued the treatment in Phase I due to AEs.31 The extension trial showed a mean 45.5% reduction in LDL-C in 17 patients who completed Week 126, which remained constant for a median of 5.1 years (range: 2.1–5.7 years).32 During this Phase, 74% patients achieved LDL-C levels below 100 mg/dL and 58% patients achieved LDL-C levels below 70 mg/dL.
The Lomitapide Observational Worldwide Evaluation Registry (LOWER),34 a prospective, non-interventional, multicentre and observational registry, reported data of 187 patients with HoFH with an exposure to treatment for up to 5.9 years. The results of efficacy and safety were consistent with the Phase III study, despite using a lower median dose of lomitapide than in the Phase III trial (10 mg/day). LDL-C reduction was 45.3% at Month 6 and 29.2% at Month 48 in those cases who remained on lomitapide. At any time after initiating treatment, 65.4% patients achieved LDL-C <100 mg/dL and 41.1% patients achieved LDL-C <70 mg/dL. Regarding AEs, no new safety issues were identified in comparison with the Phase III trial. Eighty-six patients (46.5%) experienced AEs leading to a dose reduction, and 43 (23.2%) discontinued treatment because of AEs, with gastrointestinal symptoms (13.5%) and an increase in transaminases (8%) being the most frequent causes. Temporary interruption or dose reduction of lomitapide resulted in the normalisation of transaminase levels and allowed treatment to continue. No patient experienced liver injury that was clinically overt or as assessed on laboratory findings by Hy’s law.
The benefit of lomitapide on CV events has not been evaluated. However, a modelling analysis showed a 23% risk reduction in mortality and 15% in major CV events for every 1 mmol/L in LDL-C reduction. Also, a delay of almost 6 years in the time to first event was determined,35 suggesting that an increase in life expectancy may be expected in these patients if treatment is started at age 18 years. Another post hoc analysis of the Phase III trial showed that almost half of patients with lomitapide reached a LDL-C level below 70 mg/dL after 2 years and fewer major CV events per 1,000 patients during the months of treatment was observed.36
Lomitapide has not yet been approved for use in the paediatric population. However, some reports of its use in children (aged 3–16 years), considered ‘urgent to treat’ because of early ASCVD or insufficient response to conventional treatment, have been recently published.37 After a median exposure time of 18.2 months and a median dose of 20 mg/day, a median 63.7% reduction in LDL-C levels from baseline to nadir was shown.
Mipomersen
Mipomersen is a second-generation antisense oligonucleotide inhibitor of apoB-100 synthesis. It binds to a target and specific mRNA sequence encoding apoB-100 and activating endoribonuclease H, which reduces apoB-100 synthesis and atherogenic lipoprotein concentration.38 Mipomersen was approved by the FDA for the management of patients with HoFH based on a HoFH Phase III trial.39 In this trial, 51 patients aged >12 years on maximum statin dose were allocated to mipomersen (n=34) or placebo (n=17). Mipomersen 200 mg once weekly reduced LDL-C by 24.7% (interquartile range: -31.6–-17.7%) compared to a 3.3% reduction with placebo at Week 26. Other atherogenic lipoproteins such as lipoprotein(a) and triglycerides were also reduced by 31% and 18%, respectively. The most common AEs were injection site reactions, resolving spontaneously in most patients; flu-like symptoms (FLS); and an increase in alanine aminotransferase, which was not always accompanied by an increase in hepatic fat accumulation.39
The long-term efficacy and safety of mipomersen in treating HoFH (n=38), with maximally tolerated LLT, was evaluated after 2 years of follow-up in an open-label trial.40 The reduction of LDL-C and apoB levels and AEs were consistent with those observed in the Phase III trial. FLS were the principal cause of discontinuation in almost half of the patients. Transaminase elevations and the increase in liver fat occurred in the first year of treatment but trended to return to baseline values during the second year.40
Paediatric experience is scarce. A post hoc analysis of seven patients with HoFH aged 12–18 years included in the Phase III trial and in the extension study41 showed LDL-C reduction of 30.8–62.0% in the three cases with mipomersen, and no changes with the placebo. In the extension trial, three of four patients who were initially on the placebo responded well to mipomersen, with a reduction in LDL-C of 26.5–42.0%. The safety profile was consistent with those seen in other Phase III trials.
Evinacumab
Angiopoietin-like 3 (ANGPTL3) plays a key role in the regulation of lipid metabolism by inhibiting lipoprotein lipase and endothelial lipase. Human genetic studies have shown that patients with loss-of-function variants in the ANGPTL3 gene have lower levels of LDL-C, triglycerides, and high-density lipoprotein (HDL) cholesterol, and have a lower risk for coronary artery disease (41%) than the general population.42 Evinacumab is a fully human, monoclonal antibody that works by binding to and blocking the function of this protein. The FDA has recently approved evinacumab-dgnb in patients with HoFH aged 12 years or older as an adjunct to other LLT.
The efficacy and safety of evanicumab in treating patients with HoFH have been evaluated in Phase II and III trials.43,44 In the Phase II proof-of-concept trial,43 nine patients with HoFH with stable and intensive LLT, including statins, ezetimibe, LDL-apheresis, PCSK9i, or lomitapide, received 250 mg of evinacumab subcutaneously and then 15 mg/kg through intravenous infusion at Week 2. LDL-C reduction at Week 4 (primary endpoint) was 49% (range: 25–90%). No patient discontinued treatment.
In the Phase III ELIPSE trial,44 65 patients receiving the maximum tolerated LLT were randomised to intravenous infusion of 15 mg/kg of evinacumab or a placebo every 4 weeks. A rapid drop in LDL-C was observed at Week 2, which was sustained until Week 24. At this point, patients randomised to evinacumab had a 47.1% reduction in LDL-C compared with an increase in 1.9% in the placebo group. Results were not affected by the type of mutation in LDLR gene. No patient discontinued the treatment because of an AE and no deaths occurred. The most frequent AE was FLS. The long-term safety and the effect of evinacumab on CV events have not been established.
NON-PHARMACOLOGICAL LIPID-LOWERING THERAPIES
Liver Transplantation
More than 70% of LDL-R are in the liver; therefore, liver transplantation (LT) is a therapeutic option in patients with HoFH because dysfunctional receptors are replaced by normal receptors from the donor. The first case of a heart–liver transplant was reported in 1984 in a 6-year-old girl with HoFH and recurrent angina pectoris.45 Her total cholesterol fell from 1,225 mg/dL to 268 mg/dL and the regression of xanthomas occurred early after surgery. A report 2 years after the transplant showed a restoration of LDL-R activity of 60% and LDL-C was reduced by 81%, reaching levels of 184 mg/dL. In this scenario, the addition of lovastatin normalised LDL-C levels.46 Since then, other cases of patients with HoFH, who underwent liver or heart–liver transplantations, have been reported.47 The important and significant reduction of LDL-C levels and the regression of xanthomas has been constant; however, the CV benefit is still unclear.47 Some cases have shown no development or no progression of coronary artery disease, while others have shown a slow regression. On the other hand, aortic valve stenosis may develop despite normalisation of lipoproteins with the transplantation.48
Several guidelines consider LT as an exceptional therapeutic option when patients do not respond to other treatments, or when these are contraindicated or are not tolerated.10,49 The European statement on HoFH3 recognises that LT is a successful therapeutic strategy for these patients; however, the risks associated with liver transplantation are considerable, and patients require long-term immunosuppressant therapy, thereby replacing one medical condition with another. Either way, if the possibility of a LT is considered, the decision should be made with the patient and parents or guardians, explaining benefits and potential harms.
Low-Density Lipoprotein-Apheresis
Lipoprotein-apheresis (LA) is a very effective therapeutic option for children and adults with HoFH, especially in severe cases where patients do not respond to conventional lipid-lowering drugs and during pregnancy.3 The ideal age for starting LA is before aortic root involvement starts, which occurs when an individual is approximately 5–6 years old, and always before age 10 years.3,12 Ideally, this procedure should be performed every week.
The use of plasmapheresis to treat HoFH was introduced in 1975 and an improvement in survival was demonstrated in the few patients who have undergone this procedure for approximately 8.5 years compared to their siblings who died untreated.50 The major limitation of plasmapheresis is its non-specificity, removing all proteins from the plasma and reducing HDL. Apheresis was improved to a more selective removal of apoB-containing lipoproteins, with minimal impact on other proteins and HDL. There are several techniques to remove lipoproteins; some separate blood cells, while others use the whole blood. All methods lower LDL-C by approximately 60% in a single procedure. The LA also removes oxidised LDL, inflammatory cytokines, fibrinogen, coagulation factors, and improves endothelial function, to prevent progression of atherosclerosis.51
A recent systematic review of 209 patients with HoFH showed a mean LDL-C reduction of 63–71% per session, according to the type of apheresis.52 This reduction was accompanied by the disappearance or regression of xanthomata in 83% of cases. Surrogate markers of CVD such as coronary artery disease or aortic stenosis showed less progression and, in a few cases, regression of the abnormalities after a median follow-up of 3.8 and 5.4 years, respectively. In patients with no CV events before starting apheresis, 14% developed a clinical event in the follow-up (14%), compared with 48% in patients who had previous CV events.
Recent data from an international registry showed that LA is effective and safe in children.53 The median percentage of LDL-C reduction per session was 71%, and the LDL-C level was lower in children who were treated with LA twice per week; however, few patients reached an LDL-C of <130 mg/dL. Xanthomas completely disappeared with LA in 45% of cases, and persistent xanthomatosis was inversely correlated with the duration of apheresis.53 The main AEs reported were hypotension due to bradykinin release, iron deficiency, nausea, abdominal pain, and vascular access problems.51-53
Some problems with LA are the need for specialised centres to perform the treatment, often far from patient’s home; cost; time consumption; and a worse quality-of-life, affecting long-term adherence and potential CV benefits.
THE AUTHORS´ PERSPECTIVES
HoFH is a serious and devastating genetic condition resulting from mutations in the LDLR gene, which produces extremely high plasma LDL-C levels from birth and is associated with an elevated risk of premature CV mortality and morbidity, especially as a result of aortic valve damage and atherosclerotic coronary disease. If left untreated, survival is not expected beyond the third decade of life. Making an early diagnosis and reducing cholesterol levels and the burden of atherosclerosis in the first years of life is critical to reduce the risk of developing coronary events and aortic stenosis.
Patients should be monitored and followed by specialists with experience in the management of this severe and rare disorder. Despite being a monogenic disease, the phenotypic expression is highly variable, according to the molecular defect. In this sense, null allele mutations are associated with a more severe phenotype than defective mutations, although patients still have a high CV risk. Statin and ezetimibe remain the basis of treatment together with a healthy lifestyle; in some countries, without access to new lipid-lowering drugs and LDL-apheresis, they are the only options of treatment. They should be started as early as the second year of life. Nonetheless, the response is limited to the degree of LDL-R activity. PCSK9i, which are available in Europe and the USA, are effective only in cases with defective mutations and are approved for use in treating HoFH in patients aged 12 years and older. Lomitapide, mipomersen, and evinacumab significantly reduce LDL-C levels through mechanisms independent of LDL-R and have shown to be effective in patients with HoFH. Experience with these drugs in children is scarce, and some concerns arise regarding long-term safety. Selective LDL-apheresis is a good option to treat children and adults who are refractory to LLT (Figure 1). The procedure should start before age 6 years to avoid aortic valve damage; however, it is a high-cost procedure that requires specialised centres and can compromise the quality-of-life of patients, affecting long-term adherence and CV outcomes.
Despite the availability of different treatments, most patients with HoFH do not reach the LDL-C goals proposed by the guidelines. All efforts should be made to reduce LDL-C levels and the burden of atherosclerosis with available treatments (Figure 2), in combination with the management of other CV risk factors that can be present in patients.
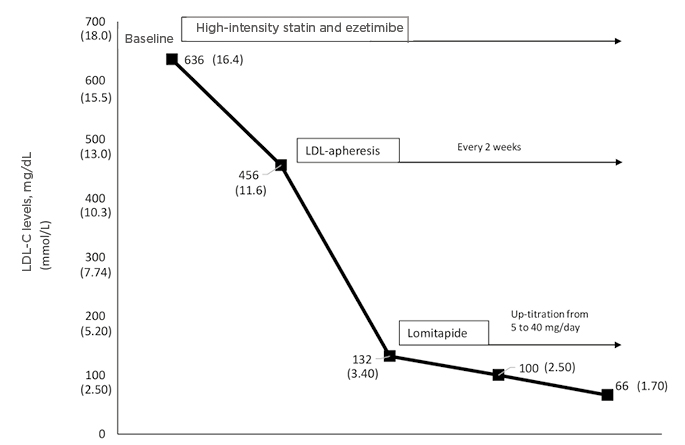
Figure 1: Response to different lipid-lowering therapies in a patient with homozygous familial hypercholesterolaemia carrying a severe mutation (null/null), showing a 90% reduction using high-intensity statin, ezetimibe, lomitapide, and low-density lipoprotein-apheresis.
LDL: low-density lipoprotein; LDL-C: low-density lipoprotein-cholesterol.
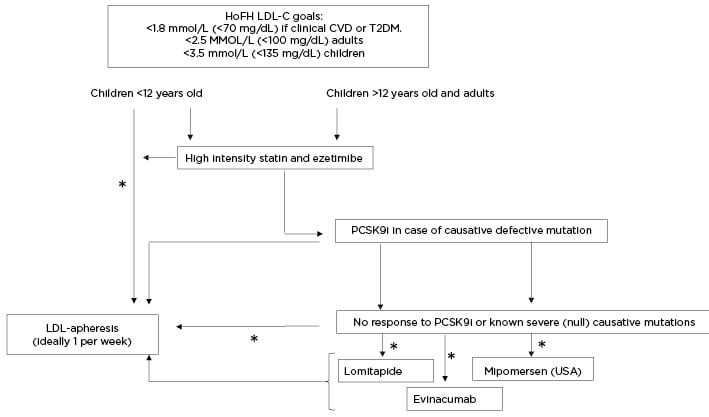
Figure 2: Clinical algorithm for the management of low-density lipoprotein-cholesterol in patients with homozygous familial hypercholesterolaemia.
*Evaluate availability, affordability, accessibility, benefits, and risks.
HoFH: homozygous familial hypercholesterolaemia; LDL: low-density lipoprotein; LDL-C: low-density lipoprotein-cholesterol; PCSK9i: PCSK9 inhibitors.