Abstract
Castration-resistant prostate cancer (CRPCa) is an advanced stage of prostate cancer in which a tumour progresses even under androgen deprivation. Treatment alternatives for CRPCa remain very limited and mostly rely on docetaxel-based chemotherapy. Despite being shown to increase patients’ overall survival, docetaxel’s clinical efficacy is impaired by development of chemoresistance. Most patients do not respond to docetaxel treatment and even those initially responsive ultimately develop resistance. Recently, chemoresistance was found to be closely related to epithelial-mesenchymal transition (EMT), a process in which epithelial cells transition into a mesenchymal phenotype. In fact, EMT markers are overexpressed in prostate cancer and are correlated to a higher Gleason score. For this reason, new therapeutic strategies are being studied to inhibit this process in several cancers. However, the clinical usefulness of targeting EMT as a way to overcome docetaxel resistance in CRPCa is still questionable and suffers from some significant limitations. This review briefly summarises the most common mechanisms of EMT-induced chemoresistance and evaluates its use as a new approach to overcome docetaxel resistance in CRPCa.
INTRODUCTION
Prostate cancer is the most common non-cutaneous cancer affecting men in Europe.1 Its incidence rates vary by >7-fold throughout the continent, with the highest rates found in northern and western countries, such as Norway and France,1,2 and have been continuously increasing over the past few decades, owing to the widespread use of prostate-specific antigen testing in prostate cancer screening.3 The well-established risk factors for prostate cancer include age, race/ethnicity (e.g. African ancestry), and family history of the disease.4
Despite having an impressive 5-year survival rate of almost 100% when diagnosed in its early stages,5 prostate cancer remains the third leading cause of death from cancer in European men, preceded only by lung and colorectal cancer.2 The main reason for its high lethality is the development of castration-resistant prostate cancer (CRPCa), an advanced stage of prostate cancer in which a tumour progresses even under androgen deprivation.6 About 80–90% of the patients who receive hormonal therapy will eventually progress to CRPCa and the overall survival after tumour recurrence drastically decreases to 1–2 years.7 Treatment alternatives for CRPCa remain very limited and mostly rely on docetaxel-based chemotherapy.8 However, some patients do not respond to docetaxel treatment and even those initially responsive ultimately develop docetaxel resistance, which is considered the main reason for CRPCa high mortality.9
Recently, epithelial-mesenchymal transition (EMT) emerged as a key mediator of drug resistance in several cancers, including CRPCa.10 Due to the current urgency in developing new therapeutic options for CRPCa, this review briefly summarises the most common mechanisms of EMT-induced chemoresistance and evaluates its use as a new approach to overcome docetaxel resistance in CRPCa.
EPITHELIAL-MESENCHYMAL TRANSITION
EMT is a process in which epithelial cells transition into a mesenchymal phenotype.11 While epithelial cells are known to show apical–basal polarity and adherence to a basement membrane, mesenchymal cells are multipolar, have a spindle shaped morphology, and present motility. Thus, during this process, epithelial cells have to undergo significant transcriptional and non-transcriptional changes that reorganise their cytoskeletal architecture, resulting in altered cell shape, increased motility, and in some cases the ability to degrade extracellular matrix proteins.11 This switch is co-ordinated by a crosstalk between several intracellular signalling pathways, including Wnt, Notch, phosphatidylinositol-3-kinase (PI3K/Akt), Hedgehog, and transforming growth factor beta (TGF-β), that activate a number of transcription factors involved in the acquisition of the mesenchymal phenotype, such as SNAIL, ZEB, and TWIST.11 The most remarkable hallmark of EMT is the loss of E-cadherin adhesion molecules, concomitant with the overexpression of mesenchymal markers such of N-cadherin and vimentin.11-13
Based on its biological functions, EMT is often classified into three different subtypes.14 The first concerns its role during embryonic development. EMT is involved in several steps of embryogenesis, including embryo implantation, placenta formation, and generation of the mesoderm, which eventually gives rise to primary mesenchyme and migratory neural crest cells. These are able to dissociate from the neural fold and disperse to other parts of the embryo.14
The second EMT subtype is involved with wounding, tissue regeneration, and fibrosis. Under the influence of growth factors produced by macrophages during inflammation and tissue injury, epithelial cells can undergo EMT to originate new fibroblasts and contribute to organ fibrosis.14 Fibroblasts originating from EMT express several mesenchymal markers, such as alpha-smooth muscle actin (α-SMA), fibroblast-specific protein 1 (FSP1), and collagen Type 1.15 In fact, Iwano et al.16 demonstrated that a substantial amount of interstitial kidney fibroblasts were actually originated from tubular epithelia at the site of injury.
Finally, the third subtype is related to EMT’s role in cancer metastasis and progression. Metastasis is a multi-step process that ultimately leads to the dissemination of primary tumours and is a major cause of cancer mortality. In order to successfully colonise further tissues and form metastasis, cancer cells firstly have to acquire the capability to detach from basal membrane, migrate through adjacent tissues, and enter into the vasculature to finally reach distal sites via haematogenous or lymphatic spread.17,18 EMT is required to enable these changes in cell adhesion and motility during tumour invasion, and therefore represents a critical regulator of the initiating events of the metastatic cascade.19 For this reason, EMT involvement in metastasis and cancer progression has been extensively investigated.15
The Emerging Role of Epithelial-Mesenchymal Transition in Chemoresistance
The link between EMT and chemoresistance has been suggested for decades, since Sommers et al.20 reported in the early 1990s that metastatic breast cancer cell lines resistant to the chemotherapy drugs doxorubicin and vinblastine presented loss of E-cadherin and the expression of the mesenchymal marker vimentin. This function of EMT was, however, somewhat expected, since most signalling pathways that regulate the EMT process have long been associated with chemoresistance in a variety of human cancers.21-23
Despite the accumulating evidence of EMT importance to chemoresistance, it was not until the recent publication of two studies that the interest in this particular role of EMT emerged. In the first one, Fischer et al.24 developed a transgenic mouse model that was able to track EMT process during breast-to-lung metastasis. Using a FSP1 promoter-driven Cre recombinase, they created a mesenchymal-specific fluorescent marker that labelled breast cancer cells with a red fluorescent marker, which turns green when cells undergo EMT. Surprisingly, they found that the majority of cells within the primary tumour and the lung metastasis had not undergone EMT. However, after treatment with cyclophosphamide, it was observed that most cells that formed metastasis after tumour relapse had undergone EMT. Gene expression profile analysis revealed that these cells presented elevated expression of chemoresistance-related genes.
In the second study, Zheng et al.25 created a genetically engineered mouse model of pancreatic ductal adenocarcinoma that harboured a tissue-specific deletion of either SNAIL or TWIST, two EMT-inducing transcription factors. Despite having no effect on metastasis formation, EMT inhibition enhanced mice sensitivity to the chemotherapy drug gemcitabine, resulting in increased overall survival. Tumour cells derived from EMT-deficient mice presented elevated expression of equilibrative nucleotide transporter 1 (ENT1) and concentrative nucleoside transporter 3 (CNT3), which are known to promote gemcitabine sensitivity in human pancreatic cancer.
Mechanisms of Epithelial-Mesenchymal Transition-Induced Chemoresistance
The mechanisms that drive EMT-induced chemoresistance are yet to be fully elucidated. However, compelling evidence shows that EMT regulates at least two major mechanisms of drug resistance in human cancers. The first involves the activity of ABC transporters, a large family of transmembrane proteins responsible for ATP- dependent efflux of many cytotoxic compounds, including drugs. The best characterised ABC transporters involved in chemoresistance are ATP- binding cassette sub-family B member 1 (ABCB1), also known as P-glycoprotein, ATP-binding cassette subfamily C member 1 (ABCC1), and ATP-binding cassette subfamily G member 2 (ABCG2). These transporters are overexpressed in several human cancers and are responsible for the efflux of a wide range of chemotherapy drugs, resulting in decreased drug intracellular concentrations and ultimately lead to drug insensitivity.26 Overexpression of ABC transporters is the predominant mechanism of multidrug resistance (MDR), the major cause of chemotherapy failure,27 and EMT plays an important role in the regulation of their expression. Breast cancer cells overexpressing EMT transcription factors TWIST, SNAIL, and FOXC2 displayed increased levels of several ABC transporters and tolerated a 10-fold higher concentration of chemotherapy. Conversely, knockdown of EMT transcription factors reduced the expression of ABC transporters in these cells and restored their sensitivity to chemotherapeutic treatment. Saxena et al.28 also demonstrated that the promoter sequence of ABC transporters has binding sites for the EMT transcription factors SNAIL, TWIST, and FOXC2, enabling EMT to directly regulate their expression.28 Therefore, the overexpression of these transcription factors during EMT leads to enhanced promoter activity of ABC transporters, followed by the acquisition of MDR.28
EMT also promotes chemoresistance by inhibiting TGF-β-induced apoptosis. Controversially, TGF-β is involved in the control of both apoptosis and EMT and the choice of which way to follow seems to be closely related to cell cycle stage.29 TGF-β-induced EMT mostly occurs during G1/S phase and results in decreased TGF-β-induced caspase activity and apoptosis.30 Cells undergoing EMT also show overexpression of anti-apoptotic B cell lymphoma-extra large (Bcl-xL) and downregulation of pro-apoptotic proteins.31 On the other hand, cells at G2/M phase mostly undergo TGF-β-induced apoptosis and display high caspase activity.30 In order to evade apoptosis, SNAIL transcription factor promotes G1/S cell cycle arrest by repressing cyclin D2 transcription and increasing the expression of p21Cip1, a cyclin-dependent kinase that impairs cell cycle progression.32 Of note, SNAIL-expressing cells have a 3-fold reduction in caspase-3 activity and are resistant to TNF-α-induced apoptosis.32
EPITHELIAL-MESENCHYMAL TRANSITION IN DOCETAXEL-RESISTANT CASTRATION-RESISTANT PROSTATE CANCER
Androgen deprivation therapy (ADT) is the standard treatment option for advanced prostate cancer.8 However, tumour regression after castration is only temporary and in 80–90% of patients the tumour relapses and progresses to castration resistance.7 One of the most remarkable features of prostate cancer transition to a castration-resistant status is the aberrant signalling of the androgen receptor (AR). CRPCa cells not only continue to express AR but are characterised by the amplification and hyperactivation of the receptor, which makes them hypersensitive to low androgen levels.33 AR signalling is activated in these cells partly by intratumoural androgen synthesis, enabling tumour growth even with very low serum testosterone levels.33
Using LuCaP35 xenografts, Sun et al.34 observed that after surgical castration tumours presented decreased levels of E-cadherin and increased expression of N-cadherin, vimentin and EMT- inducing transcription factors, including ZEB and TWIST.34 It is noteworthy that castrated tumours also had increased expression of the stem cell markers WNT5a and WNT5b. Gene expression profiles of patients who had undergone ADT also revealed an increased expression of mesenchymal markers when compared to untreated patients.34 Searching for markers of castration resistance in prostate cancer, Tanaka et al.35 compared gene expression between hormone-sensitive and castration-resistant LAPC9 xenografts and also observed higher expression of N-cadherin in the resistant mice. N-cadherin expression was also progressively raised after mice castration. Likewise, while androgen-sensitive LNCaP cell line does not express this mesenchymal marker, its androgen-insensitive clone LNCaP-CL1 does, reinforcing the theory that androgen deprivation might be related to EMT.35 However, the precise molecular mechanisms that link CRPCa to EMT are still unclear.
Furthermore, when cultured in low hormone medium, androgen-sensitive LNCaP cells underwent EMT and acquired chemoresistance, while treatment with testosterone fully restored their sensitivity to docetaxel.34 Conversely, EMT was also induced by ADT conditions in androgen-independent PC-3 cells and was reversed when cells were exposed to testosterone.36 PC-3 cells that underwent castration-induced EMT also showed increased migration and invasion capabilities.36
Interestingly, ZEB1 was overexpressed in the LuCaP35 xenografts, in human prostates, and in LNCaP cells following castration.34 ZEB1 represses E-cadherin and is a key transcriptional regulator of EMT in prostate cancer. Its expression correlates directly with Gleason score and with invasiveness, and migratory abilities of prostate cancer cells.37 Taken together, these findings indicate that EMT not only increases prostate cancer progression but can actually be induced by castration, possibly by the upregulation of ZEB1 expression. Additionally, these results raise the question as to whether patients can actually benefit from being treated with docetaxel-based chemotherapy prior to ADT.
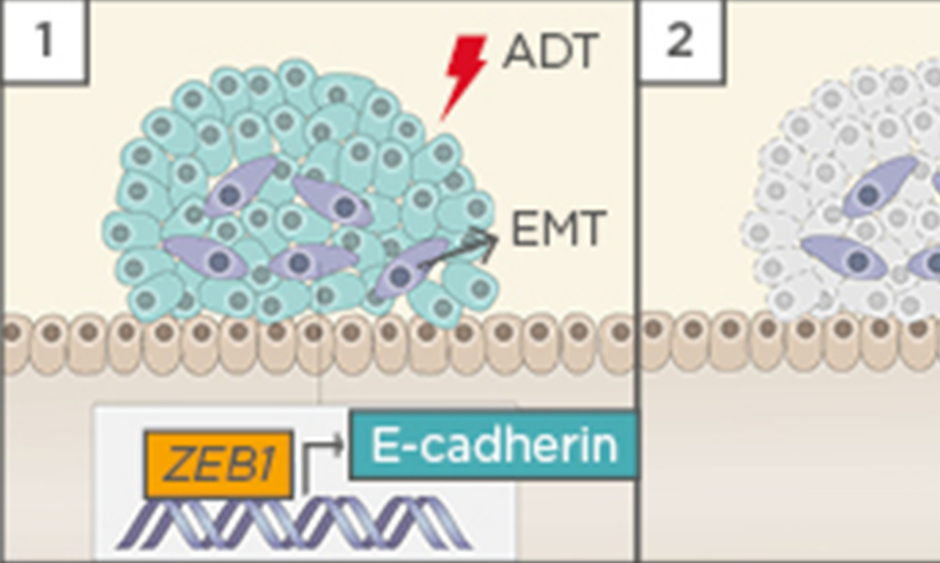
Castration-induced EMT is particularly concerning as it enables cancer cells to acquire stem cell-like features that determine chemotherapy failure.10 According to the cancer stem cell (CSC) theory of cancer, only a few niche cells, CSCs, within the heterogeneous tumour mass are capable of unlimited self-renewal, which makes them most likely to acquire mutations and to be responsible for initiating tumour development.38 Collins et al.39 discovered that a few epithelial basal cells on adult prostate tissue highly express the integrin α2β1, a stem cell marker in prostate cancer. These cells presented high clonogenic capability and were able to generate a prostate-specific differentiated epithelium in vivo when injected into nude mice. However, it is still a matter of ongoing discussion whether prostate CSCs derive from basal or luminal epithelium.40 Although the link between EMT and CSCs is still uncertain, it is known that cancer cells with EMT phenotype often display stem cell–progenitor cell-like properties. It has been reported that PC-3 cells that have undergone EMT acquired self-renewal ability by the upregulation of the transcription factors SOX2, NANOG, and OCT4, that are known to induce the reprogramming of differentiated cells to display a stem-like phenotype.41 Also, Puhr et al.42 and Marín-Aguilera et al.43 induced docetaxel resistance in castration-resistant DU145 and PC3 cell lines and observed that resistant cells had increased expression of both mesenchymal and stem-cell like markers. The expression of EMT genes also correlated with worse clinical outcome and shorter time until tumour recurrence in patients that received docetaxel treatment,43 supporting the idea that targeting EMT might be a potential strategy for overcoming docetaxel resistance in CRPCa to increase patients’ survival. The proposed model of castration-induced EMT is summarised in Figure 1.
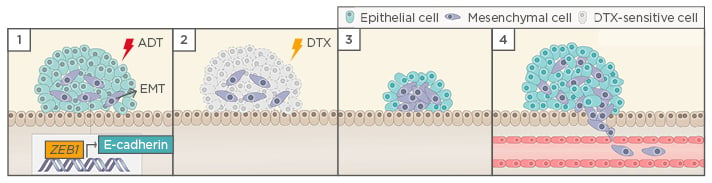
Figure 1: Hypothetical model of castration-induced epithelial-mesenchymal transition (EMT).
1) ADT triggers EMT in prostate cancer cells, possibly by the upregulation of ZEB1 expression; 2) When the tumour relapses, cells that underwent EMT after castration acquired stem cell-like features and become resistant to docetaxel-based chemotherapy; 3) EMT remanescent cells proliferate, causing tumour relapse; and 4) EMT enable cells to leave the primary tumour, migrate through adjacent tissues, enter into the vasculature, and form metastasis.
ADT: androgen deprivation therapy; DTX: docetaxel.
Targeting Epithelial-Mesenchymal Transition in Docetaxel-Resistant Castration-Resistant Prostate Cancer
So far, EMT-targeting strategies have mostly relied on the inhibition of its related transcription factors and signalling pathways. For instance, Hedgehog signalling blockade by GDC-0449 antagonist induced apoptosis and reversed docetaxel resistance of PC3 cells.44 Combined treatment with GDC-0449 and docetaxel also showed greater anti-tumoural growth inhibitory effect on PC3 cell xenografts.44 Likewise, inhibition of PI3K/Akt, Notch, and Hedgehog signallings increased docetaxel sensitivity and promoted cell apoptosis in prostate cancer resistant cells.45,46 Another target of particular interest is human macrophage inhibitory cytokine-1 (MIC-1), a member of the TGF-β protein family. MIC-1 expression progressively raises as prostate cancer progresses to metastatic state and correlates to the acquisition of EMT features and docetaxel resistance in PC3 cells.47-50 MIC-1 knockdown in these cells increased docetaxel cytotoxic effects by promoting both mitochondrial and caspase dependent apoptosis.50 Recently, Kawamura et al.51 demonstrated that CRISPR/ Cas9-mediated gene knockout of NANOG genes in DU145 cells significantly increased docetaxel sensivity. NANOG genes are major regulators of stem cells pluripotency and positively regulate the EMT process via STAT3-induced SNAIL activation.51,52 NANOG is overexpressed in prostate cancer and is associated with acquisition of stem cell characteristics, increased Gleason score, and poor prognosis.51,53 Of note, inhibition of ZEB1 in docetaxel-resistant lung cancer cells also significantly enhanced chemosensitivity.54
Another approach is to reverse the mechanisms of EMT-induced chemoresistance. ABCB1 is frequently involved in acquired docetaxel resistance in CRPCa and is upregulated in docetaxel-resistant cell lines.55 ABCB1 shRNA-mediated knockdown was able to sensitise castration-resistant C4-2B prostate cancer cells to docetaxel treatment.55 The flavone apigenin, a natural inhibitor of ABCB1 expression, also efficiently restored docetaxel sensitivity in these cells and might be considered as a natural source to modulate MDR in CRPCa. Recently, the anti-androgens abiraterone acetate and enzalutamide were shown to inhibit ABCB1 activity and therefore reduce its efflux activity.56 Combined treatment with both drugs sensitised DU145 cells to docetaxel and might be considered as a promising strategy to overcome chemoresistance in CRPCa.56 However, it is noteworthy that polymorphisms in ABCB1 largely affect the efficacy and toxicity of ABCB1 inhibitors and currently represent a significant barrier towards their clinical use.57
Finally, it is noteworthy that microRNA (miRNA)- based cancer therapy has become a research field of growing interest. miRNAs are small non-coding RNAs that control post-transcriptional regulation of gene expression. Some miRNAs are known to be involved in EMT regulation in several human cancers through the regulation of the expression of epithelial and mesenchymal markers, transcription factors, and signalling pathways that promote EMT.58 While some miRNAs exert oncogenic activity by promoting EMT, others act as tumour suppressors by inhibiting it.58 Several miRNAs related to EMT that are downregulated in CRPCa are being investigated as novel therapeutic targets, most notably miR-205 and miR-200 families. Transfection with miR-200c and miR-205 in PC-3 and DU-145 cells, which inherently express low levels of both miRNAs, resulted in restored E-cadherin expression and reduced migration and invasive capabilities.42,59 Furthermore, miR-205 induced the actual reversal of EMT, a process known as mesenchymal–epithelial transition (MET), in DU-145 cells, through regulation of genes involved in epithelial organisation and cell-cell adhesion, such as ZEB.59 Moreover, transfection of miR-205 in the docetaxel-resistant WPE1-NB26 prostate cancer cell line was able to reduce chemoresistance and enhance docetaxel- induced apoptosis,60 providing encouraging evidence on the use of nanotechnology-based strategies for CRPCa treatment.
CONCLUSIONS AND FUTURE DIRECTIONS
EMT has recently emerged as a promising topic on chemoresistance research and several strategies to impede this process are being studied as potential pharmacological approaches against chemoresistance in a broad range of human cancers, including CRPCa. However, the clinical efficacy of targeting EMT as a way to overcome docetaxel resistance in CRPCa is still questionable and presents with some significant limitations. Firstly, it is highly debatable that the simple inhibition of a specific target would successfully suppress a dynamic process such as EMT, which is regulated by multiple genes and intracellular signalling pathways. Instead, it is more likely that the inhibition of one target would be easily compensated by the activity of another EMT regulator, that exerts similar or redundant functions. Moreover, it should be considered that EMT inhibitors might exert limited benefits in advanced patients that have already undergone this transition. Patients with CRPCa would perhaps benefit more from therapies focussed on reversing EMT, therefore inducing MET, rather than inhibiting it. Instead, targeting EMT might perhaps be a more suitable approach for patients that have recently undergone ADT, as a way to prevent the progression to CRPCa and chemoresistance development. Finally, another concern relates to thesafety and clinical use of EMT targets, which is yet to be investigated. Despite these limitations, EMT remains a promising target among the current lack of therapeutic strategies for CRPCa treatment and may become clinically suitable upon further research.