BACKGROUND AND AIMS
Urothelial carcinoma in situ (CIS) of the bladder is an intra-epithelial, high grade, malignant neoplasm characterised by flat (non-papillary) growth, with high probability of disease progression.1,2 CIS has histologic features similar to invasive cancer and is often a precursor to and associated with invasive cancer. Molecularly, bladder cancer can be broadly categorised into luminal and basal subtypes.1-7 However, the molecular features that are unique to CIS, as compared to other high-grade lesions, including papillary Ta and T1 tumours, are underexplored.
Bulk RNA expression profiling is a standard for molecularly characterising bladder cancer, and RNA-sequencing protocols utilising formalin-fixed paraffin-embedded (FFPE) tumours are well established. However, CIS presents challenges for RNA sequencing due to the inability to reliably detect, the low incidence rate, and the quality of RNA derived from FFPE samples. While enhanced cystoscopy and narrow band imaging have improved the ability to detect CIS, sampling acquisition techniques still limit the optimisation of RNA sequencing. Furthermore, the authors determined that established dual nucleic acid extraction protocols for FFPE samples were not feasible due to small biopsy sample sizes, and that protocols should be optimised for either RNA or DNA. This study attempts to overcome above stated challenges to understand the molecular landscape of CIS by contrasting against papillary tumours and normal urothelium using RNA-sequencing on FFPE samples, as well as whole exome sequencing, and immunohistochemical and immunofluorescent analyses, with an intent to identify unique molecular signatures associated with CIS.
MATERIALS AND METHODS
The authors performed whole transcriptome profiling with RNA sequencing of FFPE specimens from 15 CIS, nine high-grade papillary Ta/T1 tumours, and eight normal urothelial samples (Cohort A). Wilcoxon test was used to filter differentially expressed genes and The Cancer Genome Atlas (TCGA) single sample classifier was used to assign molecular subtypes. Whole exome sequencing was performed for 19 patients with matched CIS and papillary tumour samples (Cohort B). Using multiplex immunofluorescence and immunohistochemistry analyses, 24 samples from 15 patients were analysed for the presence of cytotoxic T cells, T helper cells, regulatory T cells, B cells, M1 and M2 macrophages, and programmed cell death protein 1 and programmed death-ligand 1-expressing cells.
RESULTS
The authors performed molecular subtyping applying the UROMOL classification and as previously shown for CIS, the majority were Class 2a and 2b, with four Class 3 and one Class 1. They applied the TCGA single patient classifier and the majority were luminal with a breakdown of two luminal, five luminal infiltrated, seven luminal papillary, three basal, and one neuronal subtype (Figure 1). A 46-gene signature of differentially expressed genes in CIS samples was identified and included known druggable targets that were selectively upregulated (MTOR, TYK2, AXIN1, CPT1B, GAK, and PIEZO1) or downregulated (BRD2 and NDUFB2; p<0.05). An independent dataset was used to assess the robustness of these markers. High expression of MTOR, GUSBP11, KMT2D, and URB1 was significantly associated with CIS in this independent dataset.
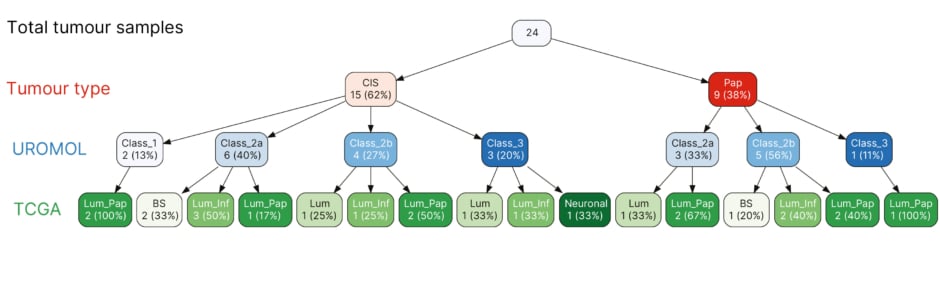
Figure 1: The Cancer Genome Atlas and UROMOL subtyping classifier (N=24; 15 carcinomas in situ versus 9 papillary tumours).
BS: basal; CIS: carcinoma in situ; inf: infiltrated; lum: luminal; pap: papillary; TCGA: The Cancer
Genome Atlas.
Additionally, mutational analysis of 34 matched CIS and 33 papillary tumours revealed a clonal origin of the lesions with mutations shared between both synchronous and metachronous tumours. Inter- and intra-patient mutational heterogeneity was also observed. The most frequently mutated gene was KDM6A, which was observed in 53% of the patient samples. Analysis of the immunological landscape of 24 CIS and papillary tumour lesions showed higher levels of immune cells in stromal compartments compared to carcinoma regions. Furthermore, more programmed cell death protein 1 positive cells were observed in CIS lesions compared with papillary tumours (p=0.03).
CONCLUSION
Collectively, this study identifies a molecular signature that distinguishes CIS lesions from papillary tumours in terms of gene expression levels, mutational landscape, and proportion of programmed cell death protein 1 positive cells that may contribute to an aggressive feature of progressive disease phenotype of the bladder cancer.