Abstract
Systemic sclerosis (SSc) is a systemic autoimmune condition of unknown cause. Yes-Associated Protein/Tafazzin (YAP/TAZ) are transcriptional coactivators previously demonstrated to be involved in cellular stretch biology, and form the principal effector molecules of the Hippo signalling pathway. The association between YAP/TAZ and stretch is contingent upon their cytoplasmic localisation (with nuclear translocation, the cell adopts a relaxed state). The author weighs the evidence for a central role for YAP/TAZ signalling in scleroderma spanning the major clinical features of the condition. Several of the features unique to SSc are mediated by cytoplasmic localisation of YAP/TAZ, including the stretch phenotype (through binding to NF-2), arterial lumenal obliteration (through their binding to angiomotin), the promotion of hypergammaglobulinaemia (via feedback to the upstream Hippo signalling molecule Mammalian Ste20-like Kinase 1), and the induction of B-Lymphocyte-Induced Maturation Protein-1 leading to the adoption of Th2 lineage, prominent in SSc. One observes that the induction of the fibrotic phenotype of scleroderma is mediated through GLI1/GLI2 (the effector molecules of the Hedgehog pathway). GLI1/GLI2 are induced to reciprocally enter the nucleus when YAP/TAZ is intracytoplasmic. The latter explains the characteristically increased connective tissue growth factor 2 and endothelin-1 expression. In this article, the author references some examples of the role of YAP/TAZ in the biophysically similar condition nephrogenic systemic fibrosis and suggests a role of YAP/TAZ cytoplasmic sequestration in programmed cell death protein 1-ligand antagonist-induced scleroderma.
INTRODUCTION
Systemic sclerosis (SSc) is a multi-system connective tissue disease characterised by excessive fibrosis, microvasculopathy, and autoimmunity. The condition broadly spans two general phenotypes, limited and diffuse, each weighted toward a characteristic but somewhat overlapping antibody and organ involvement profile.
While there has been no shortage of contributory pathogenetic component parts identified to date, a cohesive cell biological explanatory model has been elusive. In this paper, the author makes a case for Yes-Associated Protein/Tafazzin (YAP/TAZ) as the keystone in scleroderma pathogenesis.
YES-ACCOIATED PROTEIN/TAFAZZIN AND THE CANONICAL HIPPO PATHWAY
The Hippo pathway in mammals is a kinase cascade in which the mammalian Ste20-like kinases 1/2 (MST1/2) phosphorylate and activate large tumour suppressor 1/2 (LATS1/2). This cascade regulates the activity of two transcriptional coactivators: YAP and a transcriptional coactivator with the PDZ-binding motif known as TAZ. When YAP and TAZ are active, they translocate into the nucleus to bind the transcription enhancer factor 1 (TEAD 1) DNA-binding domain and induce expression of a wide range of genes that are involved in cell proliferation and in regulation of organ size. Mammals express four different TEAD (TEAD 1-4). TEAD 1-4 are broadly expressed, but each TEAD has tissue specific expression, which indicates tissue specific roles for each TEAD.1
Active MST1/2 phosphorylate SAV1 and MOB1A/B, two scaffold proteins that assist MST1/2 in the recruitment and phosphorylation of LATS1/2. Phosphorylated LATS1/2, in turn, phosphorylate and inactivate YAP/TAZ (thereby confining it to the cytoplasm). Phosphorylation of YAP and TAZ leads to their binding with 14-3-3, causing cytoplasmic sequestration of YAP/TAZ.2 LATS phosphorylation induces casein kinase 1δ/ε to phosphorylate YAP/TAZ and the recruitment of SCF-ubiquitin ligase which results in the ubiquitination and subsequent degradation of YAP/TAZ.3
As transcriptional coactivators, YAP/TAZ do not have DNA binding domains, but, upon nuclear translocation, they regulate gene expression through interaction with TEAD 1–4. TEAD 1–4 can bind to vestigial-like family member 4 (VGLL4) in the nucleus and thus function as transcriptional repressors.
The interaction between YAP/TAZ and TEAD 1-4 dissociates VGLL4 from the latter and thereby activates TEAD-mediated gene transcription to promote tissue growth and inhibit apoptosis (Figure 1).4
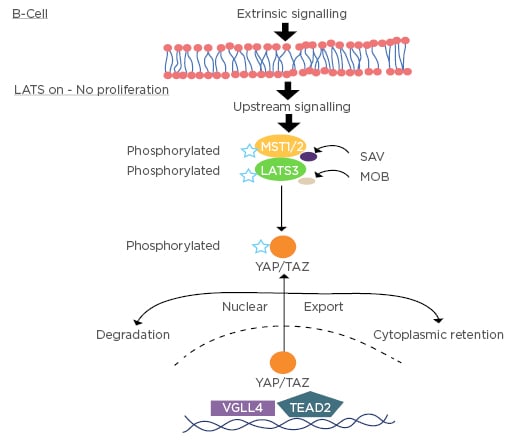
Figure 1: Hippo signalling in the B-cell–YAP/TAZ is intracytoplasmic when large tumour suppressor signalling is on.
LATS: large tumour suppressor 1/2; LATS3: large tumour suppressor 3; MST1/2: mammalian Ste20-like kinases 1/2; TEAD 2: transcription enhancer factor 2; VGLL4: vestigial-like family member 4; YAP/TAZ: yes-associated protein/tafazzin.
YES-ASSOCIATED PROTEIN/TAFAZZIN AND THE STRETCH PHENOTYPE
Upstream of the Hippo pathway are Merlin, KIBRA, and other cell adhesion molecules and polarity-regulating proteins that localise to cell–cell contact points (e.g., adherens junctions). Cell-to-cell junctions are crucial in the regulation of growth dependent control of proliferation.5,6 Merlin physically interacts with YAP/TAZ and suppresses its nuclear localisation. Cell density normally regulates YAP/TAZ nuclear localisation, thereby controlling cellular proliferation. Mechanical tension suppresses YAP/TAZ nuclear localisation via Merlin-induced nuclear export which occurs when it dissociates from the adherens junction, rendering a highly tense actin belt within the cell. Thus, the cell exhibits a tense phenotype wherein proliferation is inhibited.7 Cytoplasmic relocalisation of YAP reduces the expression levels of cyclin A, cyclin B, and CDK1 genes, both in vitro and in vivo.8 This would be anticipated to prevent progression to completion of mitosis (Figure 2).
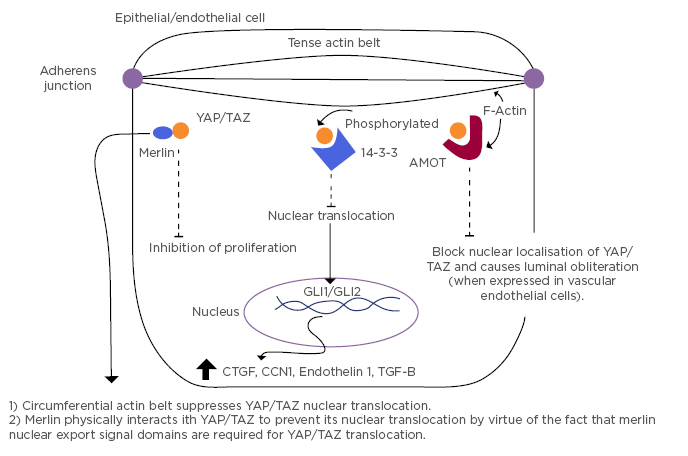
Figure 2: Mechanisms of intracytoplasmic localisation of YAP/TAZ and the resultant cytokine profile.
MOT: angiomotin; CCN1: cysteine rich angiogenic inducer-61; CTGF: connective tissue growth factor; GLI1/2: glioma-associated oncogene 1/2; TGF-β; transforming growth factor-β; YAP/TAZ: yes-associated protein/tafazzin. Adapted from Furukawa et al.7
TAZ is a component of the Wnt signalling cascade and mediates Wnt biological responses.9 When Wnt signalling is turned off, YAP/TAZ are sequestered in the β-catenin destruction complex; i.e., preventing nuclear localisation.10
RECIPROCAL EXPRESSION OF HEDGEHOG PATHWAY COMPONENTS
Another consequence of non-nuclear YAP/TAZ is enhanced expression of GLI1/GLI2 (effector molecules of the Hedgehog [Hh] pathway).11 Notably, this phenomenon was demonstrated under hypoxic conditions.
However, there is severely reduced oxygen tension in the skin of SSc patients with resultant over-expression of oxygen-regulated pathways.12 GLI1/GLI2 expression abrogates activator protein 1 (AP1) activation.13 Cytoplasmic YAP/TAZ regulates SMAD3/TGFß signalling via induction of SMAD7; however, this mechanism relies upon the AP1 transcription factor.14 Thus, it can be inferred that this SMAD3/TGFß axis is not necessarily active secondary to cytoplasmic YAP/TAZ per se. Instead, this activity may be mediated through functions of GLI1/GLI2.
Prominent over-expression of sonic hedgehog (SHH) was detected in fibroblasts of endothelial cells and keratinocytes of fibrotic skin from scleroderma patients. Expression of GLI2 was increased in the skin of patients with SSc compared with healthy controls. GLI1 was enhanced to a lesser extent. There was also evidence of prominent accumulation of GLI1/GLI2 in myofibroblasts.12
TGFß has been shown to induce transcription of GLI-2 independent of Hh proteins such as SHH, Patched (PTCH), and Smoothened (Smo). TGFß activates Hh signalling in a canonical manner via SMAD3-dependent pathways. In fact, GANT61 (a GLI2 inhibitor) ameliorated fibrosis in SSc through reduction in TGFß levels and Hh target genes.15 Hh inhibitors appear to be vulnerable to being overcome by copious IL-6 production from M2 macrophages.16
CCN MATRICELLULAR PROTEIN EXPRESSION
With reduced nuclear localisation of YAP/TAZ, there is reduced expression of connective tissue growth factor (CTGF or CCN2) and cysteine-rich angiogenic inducer-61 (CYR61, also known as CCN1). The CCN proteins are a family of extracellular matrix-associated proteins involved in intercellular signalling.17,18 CCN1 regulates keratinocyte growth and survival, as well as promoting angiogenesis, whereas CCN2 likely mediates alterations in the stromal extracellular matrix microenvironment.19
CCN1 levels diminish in avascular areas and with vaso-obliteration, whereas CTGF is induced under hypoxic conditions.20 The dearth of CCN1 expression and over-expression of CTGF/CCN2 in SSc would appear to be not entirely in keeping with YAP/TAZ cytoplasmic localisation given that presumably this results in reduced expression of both secretory proteins.17
GLI1/GLI2 are downstream mediators of TGFß (as well as effectors of TGFß transcription) and they are known to enhance CTGF/CCN2 expression,21 perhaps thereby addressing the reason for the apparently jarring finding that cytoplasmic localisation of YAP/TAZ suppresses both CCN1 (known to be suppressed in scleroderma) and CTGF/CCN2 (which is over-expressed in the condition but suppressed by cytoplasmic localisation of YAP/TAZ per se). Furthermore, overactivity of GLI1/GLI2 in capillary endothelial cells leads to intimal hyperplasia, a characteristic small vessel finding in SSc.22
KRUPPEL-LIKE FACTOR 5 AND FRIEND LEUKAEMIA INEGRATION 1 TRANSCRIPTION FACTOR
Kruppel-like factor 5 (KLF5) and friend leukaemia integration 1 transcription factor (Fli-1) have been demonstrated to be deficient in SSc.23,24
KLF5 is a transcriptional activator that binds directly to specific recognition motifs in the promoters of target genes. This protein acts downstream of multiple different signalling pathways and is transcriptionally stabilised by YAP/TAZ. It may participate in both promoting and suppressing cell proliferation. Cytoplasmic (non-nuclear) YAP/TAZ promotes proteasomal destruction of KLF5.25
Fli-1 levels are decreased in sclerodermatous lesional and non-lesional skin compared with healthy controls. There is an inverse correlation between collagen expression and Fli-1 in healthy control skin.26
To date, there is no accountable correlation between deficiency of Fli-1 expression23 and non-nuclear YAP/TAZ. However, the suppression of Fli-1 has been reproducibly demonstrated through a downstream effect of endothelin 1, the release of which is a downstream paracrine phenomenon induced by GLI1/GLI2 expression.27,28 It should also be noted that Fli-1 deficiency may contribute to depressed CCN1 levels in scleroderma.29
As highlighted by Noda et al.,30 simultaneous repression of both KLF5 and Fli-1 may be a molecular hallmark of SSc, ostensibly functioning synergistically in the pathogenesis of the fibrosis, vascular changes, and over-expression of CD19 that are characteristic of the disease.
FIBROSIS LOOPS
The Autotaxin (ATX)/lysophosphatidic acid (LPA)/IL-6 amplification loop is a driver of fibrosis in SSc, such that ATX mRNA expression was increased 2.6-fold in sclerodermatous skin as compared with healthy skin, and IL-6 expression was raised relative to the skin of controls. Sclerodermatous fibroblasts produced significantly more IL-6 in response to LPA than did control fibroblasts.31 YAP/TAZ are effector agents for the activity of LPA via LATS kinase inhibition and therefore dephosphorylation promotion, thus localising YAP/TAZ to the nucleus.32 The latter, in turn, appears to induce Merlin transcription and angiomotin accumulation, both of which will be inclined to sequester YAP/TAZ in the cytoplasm, as least in physiological circumstances.33
M2 MACROPHAGE POLARISATION
The signalling protein Wnt5a enhances TGFß1-mediated macrophage polarisation and kidney fibrosis by inducing nuclear expression of YAP/TAZ. Verteporfin-induced inhibition of YAP/TAZ was sufficient to block Wnt5a and TGFß M2 macrophage polarisation. As such, nuclear localisation of YAP/TAZ is crucial for establishing the M2 macrophage phenotype in SSc.34 Inhibition of phosphodiesterase 4 (PDE4) reduces dermal fibrosis by interfering with the release of IL-6 from M2 macrophages.35 This effect would appear to be mediated through cAMP signalling. PDE4 inhibitors have all been shown to induce YAP phosphorylation. Thus, once again this would imply that YAP/TAZ are cytoplasmic (or not active) in SSc.36 These ostensibly contradictory data point to separate simultaneous processes that result in a final common pathway.
NUCLEAR FACTOR ERYTHROID 2 RELATED FACTOR-2
Nuclear factor erythroid 2 related factor-2 (NRF-2) is a key cellular sensor of oxidative stress that can induce transcription of cytoprotective genes (such as glutathione), thus protecting cells from excessive oxidative stress. Given that SSc patients’ fibroblasts produce high amounts of reactive oxygen species that contribute to fibroblast activation and proliferation, as well as collagen synthesis,37-39 and that the pathway is druggable, some experimental trials have been conducted. First, Wei et al.40 showed that NRF-2 knockout mice displayed an exacerbated phenotype of bleomycin-induced SSc. Then, Toyama et al.41 addressed therapeutic targeting of YAP/TAZ by dimethyl fumarate (a fumaric acid ester that augments the antioxidant response by enhancing the NFR-2 signalling pathway). They demonstrated damping of TGFß1-induced gene expression through reducing nuclear localisation of YAP/TAZ and PI3K/Akt pathway inhibition.
FIBROSIS IN SCLERODERMA INTERSTITIAL PNEUMONITIS
In idiopathic pulmonary fibrosis, it was shown that mechanosignalling through nuclear localisation of YAP/TAZ is a consistent factor driving fibroblast activation and fibrosis, albeit in a small sample size (n=4 and n=5, respectively).42,43 In non-specific interstitial pneumonitis (more typical for scleroderma patients), Yeo et al.44 demonstrated that there was a more dichotomous expression of TAZ, such that there was nuclear over-expression in cellular non-specific interstitial pneumonitis (n=15) versus non-nuclear expression in fibrotic phenotypes (n=17). This may suggest a changing role for YAP/TAZ in the course of disease progression.
YES- ASSOCIATED PROTEIN/TAFAZZIN AND THE VASCULAR PHENOTYPE OF SYSTEMIC SCELEROSIS
It is notable that, at a microvascular level, the mechanotransductive properties of vascular shear stress on the endothelium results in the nuclear localisation of YAP/TAZ. In fact, the latter appears to be required for maintenance of the vessel lumen. Angiomotin and its family of proteins physically bind YAP and sequester it in the cytoplasm. Cytoplasmic relocalisation of YAP/TAZ (in this context) results in failure of lumen maintenance (i.e., obliteration of the lumen). This is a property that may at least in part be responsible for the microangiopathy component of SSc,45 perhaps borne out pathologically as capillary dropout.45-47
As previously discussed, suppression of CCN1 and KLF5, as well as the indirect effect of endothelin-148 (induced by elevated GLI2 expression), contribute to the vascular pathology witnessed in SSc.
YES- ASSOCIATED PROTEIN/TAFASSIN AND THE METABOLIC PROFILE OF SYSTEMIC SCLEROSIS
The fibrotic state promotes a glycolytic pathway of cellular metabolism. An enzymatically active pool of phosphofructokinase-1 that binds TEAD 1–4 and fosters YAP/TAZ activity (demonstrated in malignant and non-malignant cell lines). Phosphofructokinase-1 stabilises YAP/TAZ interaction with TEAD 1–4.49
TEAD 4 directly targets peroxisome proliferator-activated receptor (PPAR)-γ activity and acts as a regulator in the early stage of adipogenesis. In adipogenesis, TEAD 4 is dramatically increased and binds to promoters of adipogenic genes. TEAD 4, together with its cofactors VGLL4 and C-terminal binding protein-2 (CTBP2), negatively regulate adipogenesis. This ternary complex inhibits adipogenesis and PPAR-γ promoter activity.
VGLL4 is a competitive antagonist of YAP/TAZ at TEAD 4. Both adipogenesis and PPAR-γ expression appear to be curtailed in SSc, once again insinuating cytoplasmic localisation of YAP/TAZ.50,51 Lats2, which supresses preadipocyte proliferation (characteristic for SSc), enhances the phosphorylation and cytoplasmic accumulation of YAP/TAZ. Notably, PPAR-γ is also induced by KLF5, the expression of which is stimulated by C/EBPß.52 Korman et al.53 demonstrated that pharmacological restoration of PPAR-γ signalling may help to control skin fibrosis in SSc.
YES-ASSOCIATED PROTEIN/TAFAZZIN AND THE IMMUNOLOGICAL PROFILE OF SYSTEMIC SCLEROSIS
CD19 is approximately 20% over-expressed in scleroderma. CD19 is a potent positive regulator of B-cell function. It potentiates signals from the B-cell receptor, which results in PI3K activation and subsequent Akt phosphorylation.54-56 CD19 is a driver of B-cell activating factor, which has been shown to be over-expressed in scleroderma.57 Furthermore, the expression of proinflammatory adipokines, such as IL-6, MCP-1, COX-2, and haptoglobin, was highly increased in B-cell activating factor-treated 3T3-L1 adipocytes, while the level of adiponectin was grossly reduced.58
MST1 (a component of the canonical Hippo signalling pathway) positively regulates B-cell receptor signalling via regulation of CD19 transcriptional levels.59 YAP/TAZ act downstream from MST1. When MST1 signalling is activated, YAP/TAZ are deactivated and/or located in the cytoplasm. MST1 moves to the nucleus and binds to TEAD 2. TEAD 2 is the specific subtype of TEAD expressed predominantly in B-cells. In binding to TEAD 2, there is interaction with the 3’ UTR of CD19 promoting CD19 transcription andconsequent translocation to the B-cell membrane. This, in turn, contributes to hypergammaglobulinaemia and autoantibody expression (Figure 3).
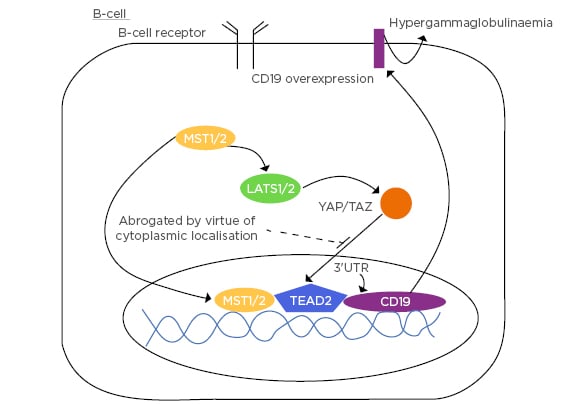
Figure 3: B-cell demonstrating the promotion of CD19 via MST1 occurring in the context of intracytoplasmic YAP/TAZ.
3’UTR: 3’ prime untranslated region; LATS1/2: large tumour suppressor 1/2; MST1/2: mammalian Ste20-like kinases 1/2; TEAD2: transcription enhancer factor 2; YAP/TAZ: yes-associated protein/tafazzin.
MST-1 is fundamental to T-lymphocyte function from thymocyte migration and antigen recognition, through to shaping the adaptive immune response.60
The degradation of YAP contributes to higher expression of B-lymphocyte-induced maturation protein-1 (BLIMP-1), which mediates the terminal differentiation of CD8+ T cells.61 CTLA-4 activation regulates the expression of BLIMP-1 in CD8+ T-cells by activating (i.e., facilitating nuclear transfer) of YAP/TAZ,62 a mechanism which may underlie the clinical efficacy of abatacept (a CTLA-4 agonist) in clinical trials of scleroderma treatment.63 BLIMP-1 expression is mainly in activated T cells and is essential for the production of IL-10 by a subset of Foxp3+ regulatory T cells with effector phenotype.64 Previous studies have demonstrated an important role for IL-10 in skin and pulmonary involvement in SSc.65,66
BLIMP-1 promotes Th2 cell differentiation.67 Th2 cells secrete IL-4, IL-5, IL-9, and IL-13, and are the predominant T-cell phenotype in scleroderma.68 Notably, Hh signals have also been shown to promote Th2 differentiation.69
In developing plasmablasts, BLIMP-1 is dispensable for IL-10 production (interferon regulatory factor 4 is necessary), and B cells lacking BLIMP-1 fail to fully differentiate into plasma cells.70 Early expression of BLIMP-1 in B-cells in fact causes autoimmune disease by inducing an increase in self-reactive plasma cells.71
To date, the YAP/TAZ cascade does not explain the specific antibody profile of SSc patients (anti-topoisomerase, anti-centromeric antibodies, anti-RNAP-3). The common feature of these antibodies would appear to be the fact that these are both directed against cell-cycle progression elements. As such, these may constitute features of a response to induced cellular senescence.
YES-ASSOCIATED PROTEIN/TAFAZZIN IN SCLERODERMA: PHENOMENON OR EPIPHENOMENON?
Monocytes from African American SSc patients with interstitial lung disease have been shown to have low levels of caveolin-1 leading to preferential fibrocyte differentiation.72 Caveolin-1 and YAP are positively regulated (i.e, when YAP is intracytoplasmic, caveolin-1 expression is low).73
Nephrogenic systemic fibrosis is a condition that bears some similarity to scleroderma. The condition was first identified in 1997 among individuals on dialysis/renal patients who had exposure to gadolinium-based contrast agents used for MRI. The condition is characterised by tightening of the skin with resultant joint contractures. Pathologically, it differs from scleroderma insofar as there is a proliferation of fibroblasts and deposition of elastic fibres and thickened collagen bundles (closely resembling the pathology of scleromyxedema). The specificity of this finding is due to the relatively prolonged exposure to gadolinium (Gd3+) ions, due to the failure of the kidneys in these individuals to excrete the ions.74-76
Gd3+ binds to and inhibits PIEZO-1,77 a cell-surface-expressed mechanotransductive (stretch activated) calcium channel critical to homeostasis of sheets of epithelial cells in vivo. Where stretch of epithelial cells occurs, as in a sheet of epithelial cells, to the extent of 1.6 psi (phi, or the golden ratio), PIEZO-1 transduces a signal that results in cell division.78 In the absence of this signal, the sheet of epithelial cells enter a ‘tense’ state wherein they are unable to compensatorily divide sufficient to accommodate ‘normal’ movements.77,79
Specifically, to affect this state, PIEZO-1 activation has been demonstrated to cause decreased nuclear localisation of YAP/TAZ.80 While the pathology of nephrogenic systemic fibrosis and SSc are not the same, both conditions are undeniably biophysically similar.
It has been demonstrated recently that the immune checkpoint inhibitor and PD1 antagonist, pembrolizumab, may induce a condition indistinguishable from scleroderma.81 The significance of this is that when leucocyte-associated PD1 binds to the target cell-associated PD1 ligand (PD-L1), cytoplasmic YAP/TAZ acts downstream of the PD-L1 by translocating to the nucleus, binding TEAD, and thereby promoting further PD-L1 proliferation.82,83 Notably, other pathways also promote PD-L1 proliferation by encouraging nuclear localisation of YAP/TAZ. By blocking the PD1/PD-L1 interaction, no downstream signal is sent to the cytoplasmic YAP/TAZ and thus one may reason that an excess of YAP/TAZ will remain intracytoplasmic. The consequence is that the phenotypic features of scleroderma are expressed. Further study of checkpoint inhibitor-induced scleroderma is necessary to confirm the role of YAP/TAZ in this phenomenon.
CONCLUSION
It remains difficult to parse the exact aetiology of SSc. Part of this difficulty is rendered by the substantial overlap in cytokine and kinase pathways. Fibrosis is a final common pathway of many inflammatory processes and, as such, looking through the prism of established disease distorts underlying causative mechanisms. There are some studies that imply or categorically argue in favour of nuclear localisation of YAP/TAZ in scleroderma. While this may be a consequence of other potent cytokine influences in established disease, this would not make logical sense initially since this would imply a ‘relaxed’ cellular phenotype with pro-proliferative tendency.
There is conflation too at the level of interpreting the reality of a ‘stiff’ microenvironment (as seen with a fibrotic extracellular matrix) and a ‘stretched’ cellular phenotype. The reality is that both coexist in vivo in scleroderma. This is a challenging model to recreate per se, let alone to evaluate its functional aberrancies in a diseased state.
There is a weight of evidence supporting a substantive role for hippo pathway mediators in SSc encompassing all the major features of the condition. Most features favour intra-cytoplasmic localisation of YAP/TAZ. There is apparent functional desynchrony of YAP/TAZ, GLI1/GLI2, Wnt pathyway components, TGFß, and intracellular oxidants (Piersma B et al.84 and Korman B et al.85 have provided excellent reviews on this subject). This ultimately results in a disease that is grossly refractory to current treatments or cures. Knowing this, careful focus should be applied simultaneously to pathway blockade mechanisms in disease treatment and to further elucidate aetiological mechanisms.