Abstract
Rheumatic diseases collectively are complex disorders, often with multifactorial origins ranging from genetic risk factors to viral triggers. In many cases, the exact pathogenic mechanisms are poorly understood. Treatment response is often difficult to predict, and significant research is currently being undertaken to investigate new avenues for potential novel therapies. Immunometabolism, the study of the interface between immunological and metabolic processes, represents one such avenue at the forefront of this research and links cellular metabolism with the various changes in immunophenotypes observed across a variety of rheumatic disorders. Abnormal mitochondrial function and dysregulation of energy metabolism has been proposed as a potential mechanism for the pathogenesis of systemic lupus erythematosus, inflammatory arthritis, and vasculitis. Furthermore, various metabolomic and amino acid changes have been observed across rheumatic diseases during activation of the immune and inflammatory response, thus representing an attractive prospect for medication development. In this review, the authors focus on immunometabolism in rheumatic disease, looking at mitochondrial dysfunction, fatty acid metabolism, and protein and amino acid changes across the disease spectrum. In particular, the authors evaluate the implications for the understanding of disease pathogenesis and explore the potential for immunometabolic intervention as a means of treatment.
Introduction
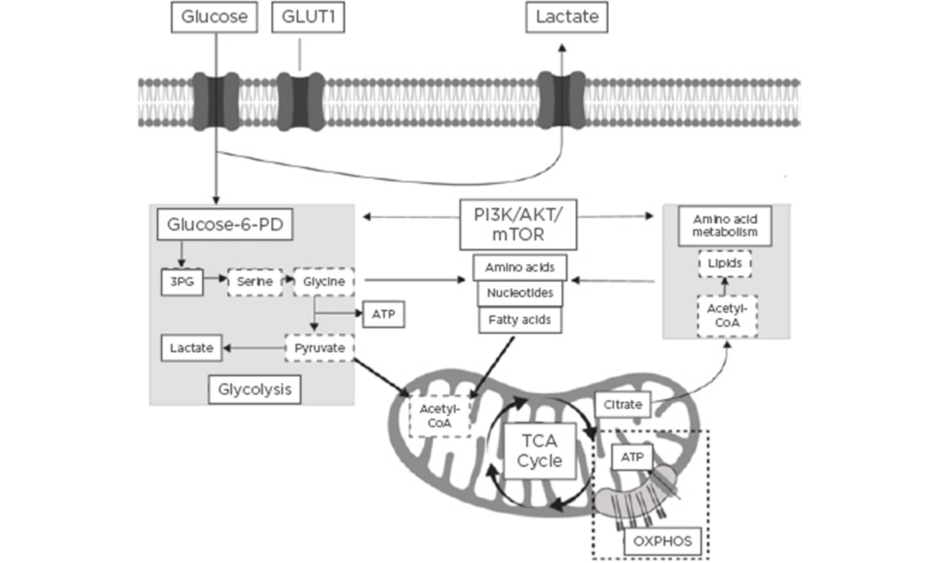
The field of immunometabolism is a rapidly expanding area of research that centres around understanding the interrelationship between immunological and metabolic processes.1 Activation of the immune system is a dynamic process that requires significant immune metabolic reprogramming to induce and maintain proliferation of immune cells, as well as activation and engagement of effector cellular function.2 Immunometabolism encompasses the roles of glycolytic and mitochondrial-derived energy metabolism, regulation of fatty acid oxidation and lipid synthesis, and protein kinase and amino acid metabolism.3 A summary of the key immune cell metabolic pathways is highlighted in Figure 1.
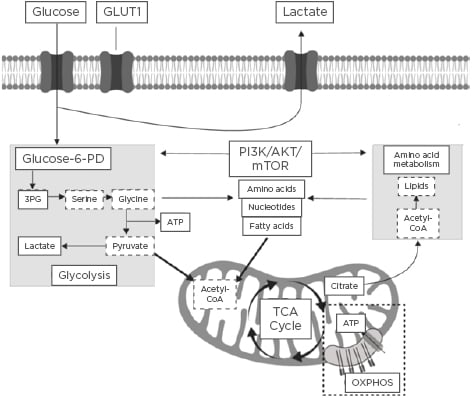
Figure 1: Summary of the key immune cell metabolic pathways.
In recent years, significant research has shed new light on the pathogenesis of various diseases with abnormal immunometabolism, including the development of atherosclerosis,4,5 diabetes,6,7 multiple sclerosis (MS),8 and malignancy.9 This has resulted in further research investigating the potential to alter the immunological–metabolic interface, which may represent a possible novel route towards new therapeutic targets.
Autoimmune rheumatic diseases are associated with activation of both the innate and adaptive immune system and results in the generation of autoantibodies and pro-inflammatory cytokines. This heterogenous group of disorders are typically characterised by a number of shared pathological mechanisms with a variety of different immunometabolic pathways implicated.
In this review, the authors describe the role of metabolic pathways during immune activation, evaluate the latest evidence supporting the role of changes in immunometabolism in various rheumatic diseases, and consider how this may lead to potential novel future therapeutic options.
Glycolysis, Mitochondria, and Energy Metabolism
Cellular metabolism is dependent upon two key metabolic pathways that are required to produce energy in the form of adenosine triphosphate (ATP): glycolysis and oxidative phosphorylation. In health, glycolysis is the metabolic pathway that converts glucose to pyruvate and hydrogen ions, which are essential for ATP generation. In the context of immune cell activation, metabolic reprogramming in glycolysis pathways are required for the induction and maintenance of cellular proliferation. Macrophages activated by lipopolysaccharide (LPS) have been demonstrated to switch their core metabolism to the glycolysis pathways. However, this change in metabolism pathways has been associated with an accumulation of a number of Kreb cycle intermediates, such as succinate, which stimulates IL-1β production and may induce a pro-inflammatory state.10 Furthermore, metabolites, including fumarate and itaconate, have been implicated in this adaptive immune response.11,12
In the pathogenesis of autoimmune rheumatic diseases, glycolytic pathways have been studied in the context of autoreactive T cells in systemic lupus erythematosus (SLE), which are dependent upon glycolysis for early inflammatory effector functions. The activity of calcium/calmodulin-dependent protein kinase 4 has been suggested to be responsible for glycolytic pathways and, in turn, contributes to aberrant expression of the GLUT1 receptor in active SLE.13 Yin et al.14 previously demonstrated that by normalising T cell metabolism through inhibition of glycolysis with 2-deoxy-d-glucose, interferon-γ production in a murine SLE model was reduced. Whilst glycolysis has been implicated in the initial immune response, T cells that become chronically activated predominantly generate ATP from mitochondrial oxidative phosphorylation rather than glycolysis.15
In comparison to glycolytic energy metabolism, mitochondria produce ATP through oxidative phosphorylation using oxygen and nutrients, which is driven via an electrochemical gradient along the inner mitochondrial membrane. Mitochondria also represent the major source of reactive oxygen species (ROS) generation. A number of studies have observed that mitochondria can be potent activators of the immune-mediated inflammatory response.16,17 In recent years, the study of mitochondria dysfunction, altered bioenergetic conditions, and ROS production have been investigated in the pathogenesis of a number of rheumatic diseases.
Mitochondria contain their own genetic material in the form of mitochondrial DNA (mtDNA) and this has also been implicated in the pathogenesis of various rheumatic diseases. Impaired energy metabolism can induce mitochondrial hypoxia, which has been shown to cause point mutations in mtDNA taken from the synovial tissue of patients with inflammatory arthritis. Further research showed the addition of antioxidants (in this case N-acetylcysteine [NAC]) rescue these mutations.18 In addition, effective treatment with anti-TNF-alpha therapy has been demonstrated to reverse these mtDNA mutations.19 Mitochondrial dysfunction results in damage to the structure of the organelle and ultimately in the release of mitochondrial genetic material from the cell into the microenvironment. This circulating cell free mtDNA can be detected in plasma and has been implicated in the pathogenesis of granulomatosis with polyangiitis (GPA), in which levels of mtDNA were found to be significantly elevated in those who were untreated, suggesting this may be a potentially novel biomarker.20
There is growing evidence from a number of studies supporting the role of abnormal mitochondrial function in the pathogenesis of osteoarthritis (OA). OA chondrocytes stimulated by IL-1β have been noted to demonstrate high levels of ROS generation and mitochondrial membrane damage, which has been associated with a higher incidence of apoptosis.21 Rheumatoid arthritis (RA) synovial fibroblasts have also demonstrated significant mitochondrial dysfunction and abnormal autophagy, which has also been seen in chondrocytes derived from patients with OA.22,23
The role of oxidative stress in the pathogenesis of SLE is well described,24 however, the implications for mitochondrial dysfunction resulting in ROS production in the pathogenesis of the disease is a more recent area of interest.25 Mitochondrial electron transport chain complex one has been reported as the main source of oxidative stress in peripheral lymphocytes in SLE. Furthermore, it was noted that NAC inhibited ROS production and proposed that this may be of possible therapeutic benefit.26 Mitochondrial ROS have also been found to induce the formation of neutrophil extracellular traps (NET) through NETosis,27 which has been implicated in the development of various autoimmune rheumatic disorders. A study by Lood et al.27 found that mitochondria-derived ROS are essential for the induction of maximal NETosis in SLE. The authors also noted that inhibition of ROS formation in vivo resulted in a reduction in both type I interferon signature and disease severity in murine models. It was concluded that both NET and pro-inflammatory oxidised mtDNA play a key role in the pathogenesis of SLE.27 Figure 2 summarises the ways in which mitochondrial dysfunction can result in ROS generation, NETosis, and the release of mtDNA. Previous animal studies have also implicated NETosis in the development of antibody-mediated thrombosis in antiphospholipid syndrome.28,29 Mitochondrial oxygen consumption was also noted to be elevated in the liver of 4-week-old lupus-prone mice, which led to the formation of anti-phospholipid antibodies prior to the onset of the disease phenotype. Furthermore, this was observed to be corrected with the addition of rapamycin,30 a drug that targets and modulates autophagy pathways.
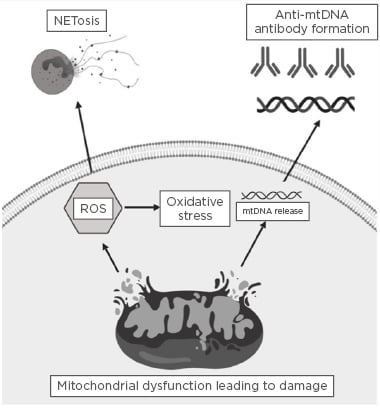
Figure 2: Summary of how mitochondrial dysfunction can result in reactive oxygen species generation, NETosis, and the release of mitochondrial DNA.
Mitochondrial dysfunction can induce damage to the outer mitochondrial membrane, which in turn can lead to the generation of reactive oxygen species and induce oxidative stress. Significant mitochondrial damage can result in mitochondrial DNA release, and, in turn, this circulating antigenic mitochondrial DNA may result in the formation of autoantibodies directed against mitochondrial genetic content. mtDNA: mitochondrial DNA; ROS: reactive oxygen species.
Lipid Metabolism
Lipids are a critical aspect of metabolism, playing fundamental roles in cell membrane composition, membrane receptor signalling, and energy storage. The key lipids for cellular function include cholesterol, phospholipids, fatty acids, triglycerides, and glycosphingolipids (GSL). Lipid metabolism is implicated in a wide range of diseases including cardiovascular disease (CVD) and nonalcoholic fatty liver disease; however, more recent studies have shown a significant role for lipids in regulating inflammation and driving autoimmune diseases.31-33
Lipid metabolism is used in different ways depending on the immune cell. For example, regulatory T cells (Treg) use lipids for their anti-inflammatory functions through beta-oxidation in the mitochondria and generate ATP through oxidative phosphorylation, whereas effector T cells depend more highly on glycolytic over lipid mediated processes for the growth and proliferation necessary for their functions.31 Lipids also play a significant role in the immune cell membrane in signalling platforms called lipid rafts.32,33 These comprise signalling proteins, GSL, and cholesterol, which together mediate T cell and B cell receptor signalling through co-receptor recruitment to the raft.
CVD is a major complication of autoimmune diseases and this is largely due to prolonged inflammation and dyslipidaemia.34 Dyslipidaemia broadly relates to the disrupted balance between low-density and high-density lipoproteins (LDL and HDL), which are pro-atherogenic and anti-atherogenic, respectively. Lipoproteins are responsible for transporting processed lipids, such as cholesterol and triglycerides, to HDL and from LDL in the liver.35 SLE is a common example of an autoimmune disease heavily influenced by dyslipidaemia, and CVD has been shown to be the leading cause of mortality for SLE,36,37 largely due to atherosclerosis. During atherosclerosis, macrophages take up the oxidised form of LDL in arteries, eventually resulting in macrophage foam cell formation and the formation of fatty lesions in the arterial wall. Rupture of the vessel wall can occur with excessive build-up of these fatty lesions, resulting in the recruitment of platelets, thus leading to narrowing of the arterial lumen.38 It has also recently been shown that lipoproteins can control the balance of lipids in the immune cell membrane, thus controlling inflammation, another key driver of atherosclerosis.39 In addition, lipid rafts have also been shown to be disrupted in SLE.32,40 Jury et al.41,42 showed an increase in cholesterol and GSL at the membrane to increased T cell receptor signalling at lipid rafts, and that a therapeutic intervention of GSL synthesis can normalise this signalling and reduce inflammation. Cholesterol is known to be involved in T cell activation,43 thus it is another metabolic target for therapeutic agents, such as statins. Altered lipid rafts have also been shown to impact B cell receptor signalling in SLE.44 Cholesterol has also been found to play a number of roles in the activated immune response. In autoimmunity, cholesterol metabolism has been implicated in the production of IFN-γ and immune complexes,45 which have a significant role in the pathogenesis of a number of rheumatic conditions.
Similarly, despite being a disease associated with inflammation of the joints, comorbid conditions in RA have also been related to dyslipidaemia and CVD;46 however, data is conflicting.47 This is again likely to relate to the generalised effect of lipid metabolism and inflammation on early atherosclerosis in RA.34 Active RA patients have been shown to have increased circulating HDL-cholesterol, and one study has demonstrated that this is also the case for untreated patients.48 In addition, a separate study showed that smaller sizes of LDL and HDL, commonly shown to have more pro and anti-atherogenic effects respectively than their larger counterparts, were increased and decreased in the serum of RA patients, respectively.49,50
Current treatments for rheumatic disorders have been shown to influence lipid metabolism. An example in SLE is the use of hydroxychloroquine (an antimalarial agent), which has been shown to reduce levels of circulating LDL.51 In contrast, the prolonged use of corticosteroids in SLE is associated with driving further dyslipidaemia, despite its preferential effects on inflammation.52,53 In addition, RA patients treated with glucocorticoids display increased levels of HDL.54 Regarding lipid modification therapy, high dose statins (80 mg/day) are currently being trialled as a new therapy for patients with MS and the Phase II trial showed reduced rates of brain atrophy and disability progression in patients with secondary progressive disease.55,56 In addition, RA patients treated with statins have shown improvements in erythrocyte sedimentation rate and C-reactive protein compared to patients on conventional standard of care therapy after 6 months of follow-up.57 However, evidence that statins are beneficial in SLE patients, in terms of reducing cardiovascular risk and/or inflammation, is mixed. Some smaller studies have shown a beneficial effect;58-61 however, the Lupus Atherosclerosis Prevention Study62 and Atherosclerosis Prevention in Paediatric Lupus Erythematosus study63 did not identify any beneficial effects of statins on disease activity or CVD risk measurements. Follow-up analysis has identified that patients with higher baseline C-reactive protein did, however, have improved CVD risk measures following the trial.64,65 Thus, the success of future trials may depend on correct stratification of patients based on lipid profile and improved suitability of primary outcome measures.
Lipid metabolism is a key player in autoimmunity, and the therapeutic targeting of specific pathways holds promise for dual mediation of inflammation and CVD. The pathways that need to be targeted and the impact of these physiologically will need to be carefully considered, including the differential role of lipid metabolism across immune cell subsets. Further studies are required, but this opens the possibility of modulating diet to influence lipid metabolism as a potential treatment for autoimmune disease.
Protein Kinase and Amino Acid Metabolism
Proteins, peptides, and amino acids play an important role in immunometabolism in both health and disease, particularly with their effects on T cell differentiation and function. An imbalance between the pro-inflammatory T helper cell subsets, Th1 and Th17 cells, and anti-inflammatory Foxp3+ Treg, with the subsequent loss of self-tolerance, is thought to contribute to autoimmune disease.66,67 Normally T cell differentiation into various cell subsets relies on the activation of the mTOR, a serine-threonine protein kinase that is present in two different complexes: mTORC1 and mTORC2.68 It also helps maintain cell homeostasis by regulating metabolic signals and nutrient availability to drive genetic programmes involved in cell growth, activation, energy use, proliferation, and survival.68-71 Through sensing cell energy status and the available metabolites, mTOR is capable of altering cellular activity.68 mTORC1 activation alters T cell metabolism to provide the essential constituents required for Th1 and Th17 cell proliferation and differentiation; however, this signalling is also necessary for the suppressive function of Treg.71
In patients with SLE, mTORC1 activation occurs in CD4+ T cells.69 This activation of mTORC1 may be driven by mitochondrial dysfunction secondary to the depletion of the tripeptide glutathione;72 through hyperactivation of the pentose phosphate pathway and increased transaldolase activity;70,73 or by a rise in the tryptophan metabolite kynurenine, which has immunomodulatory functions.70,74 In rare cases, genetic activation of mTORC1 is possible.70,75 mTORC1 activation may also represent a biomarker of autoimmune inflammation68,69 and has been implicated in the pathogenesis of SLE in several ways. For example, activation has been detected after an increase in glycolysis and suppression of autophagy (including mitophagy) with subsequent mitochondrial dysfunction.70 T cell necrosis, decrease in Treg populations, and an increase in pro-inflammatory Th17 and double-negative (CD4-CD8-) T cells76,77 have also been observed. Double-negative T cells then in turn stimulate B cells to produce autoantibodies in SLE.77 Similarly in RA, there are metabolic interactions between enzymatic proteins in T cells, which are believed to play a key role in chronic inflammation underlying the disease.78 T cells in RA are chronically activated, and thus undergo metabolic reprogramming, ultimately existing in a state of energy deprivation.78 In early RA, this process occurs in lymphoid organs, with reduced activity of the enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) in CD4+ T cells.78,79 PFKFB3 is an enzyme that normally produces fructose 2,6-bisphosphate, which, in turn, activates the rate limiting enzyme in glycolysis, phosphofructokinase 1.67,78 Reduced PFKFB3 activity results in a decrease in glycolysis, lower pyruvate and ATP levels, and shunting of glucose into the pentose phosphate pathway.78,79 These T cells are predisposed to apoptosis and fail to induce autophagy, a process normally required for cells to recycle their internal biosynthetic precursors for energy generation.78
While shunting to the pentose phosphate pathway allows T cells to produce biosynthetic precursors required for clonal expansion, it has significant metabolic and functional consequences in RA.67 Higher levels of nicotinamide adenine dinucleotide phosphate (NADPH) and reduced glutathione are produced, which neutralises ROS.67 ROS normally act as messengers required for appropriate T cell activation, proliferation, migration, and apoptosis via oxidant signalling.67,78,79 As a result of the depletion of ROS, oxidation-dependent cell signalling becomes dysregulated and there is insufficient activation of the cell cycle kinase ATM.67 Thus, T cells become hyperproliferative and favour differentiation into pro-inflammatory Th1 and Th17 cell lineages.67,79 In the later stages of RA, T cells invade peripheral tissues, such as synovial joints, interacting with B cells, plasma cells, antigen presenting cells, and tissue resident cells to create a lymphoid structure.67,80 This lymphoid structure has hypermetabolic activity with immune cells continuing to release metabolites that promote inflammation in the surrounding synovial tissue.67
Increased understanding of these pathways could lead to more precise treatment of autoimmune rheumatic diseases because specific metabolic pathways could potentially be targeted to modify an immune cell response.81 For example, both rapamycin (also known as sirolimus) and NAC inhibit mTORC1 and decrease disease activity in SLE patients.72,82-84 A recent single-arm, open-label Phase I/II trial of 43 patients with treatment resistant and/or treatment intolerant SLE, found that disease activity improved following 12 months of sirolimus, particularly in those with mucocutaneous and musculoskeletal symptoms.84 Sirolimus decreased IL-4 and IL-17 expression by pro-inflammatory double-negative and CD4+ T cells and upregulated Treg.84 Unfortunately, the benefits of sirolimus are counterbalanced by commonly observed side effects including infection, hyperlipidaemia, and hyperglycaemia,68,85 which is a concern in SLE because infections and CVD contribute greatly to mortality.86 In comparison, NAC has few side effects72 and the rationale behind its use is based on studies suggesting that both oxidative stress and reduced glutathione play a key role in the pathogenesis of SLE via abnormal T cell activation.87,88 As well as inhibiting mTORC1, NAC reverses glutathione depletion, reduces double-negative T cell proliferation, and upregulates Treg.72 Larger randomised controlled trials are required to further evaluate the effectiveness of sirolimus and NAC in SLE and other autoimmune rheumatic disorders characterised by abnormal mTOR activation.
Conclusion
In conclusion, recent advances in the understanding of the role of abnormal immunometabolism have shed new light on the pathogenesis of a number of rheumatic diseases. Similarly, research into lipid metabolism is revealing the ways in which rheumatic diseases are associated with non-traditional mechanisms of CVD. Understanding these complex interactions between metabolism and inflammation raises exciting opportunities to develop new innovative treatment options. For example, there is the possibility of using a variety of novel therapeutic agents including antioxidants (such as NAC) or even dietary modification (as a means of changing lipid profile) to ultimately improve disease activity and reducing symptoms in the future.