Abstract
Behçet’s syndrome (BS) is a systemic vasculitis with a wide range of clinical presentations and disease courses. It may involve the mucosa, skin, joints, vessels, eyes, and nervous and gastrointestinal systems. These organ involvements may present alone or co-exist in the same patient. Three main clusters of commonly co-existing manifestations were recognised and are currently called disease phenotypes. There is a significant heterogeneity among patients regarding demographic features and clinical expression of the disease that hinders a standardised disease assessment and a generalised use of diagnostic criteria. Additionally, BS is not associated with pathognomonic laboratory or histopathology features; therefore, the diagnosis is mainly based on the clinical manifestations. The purpose of this narrative review of the literature is to provide a description of the most common or typical clinical features of BS, summarise the major phenotypes of BS, and address the diagnosis strategy of this syndrome.
INTRODUCTION
Behçet’s syndrome (BS) was first described by Hulusi Behçet in 1931, as a definite clinical entity, the ‘triple symptom complex’, based on three prominent signs: relapsing oral apthae, genital ulcerations, and iritis.1 Although these are the most frequent manifestations, considered hallmarks of the disease, it was later recognised that BS is a multisystem inflammatory condition that may involve additionally the vascular, musculoskeletal, gastrointestinal (GI), and nervous systems.2 The manifestations range from self-limiting symptoms, with unpredictable relapses, to severe clinical flares that may result in organ damage or death.
A variable vessel vasculitis is thought to underlie the clinical manifestations of the disease, although the presence of a vasculitis is difficult to discern in some of its features.3 The exact aetiology and pathogenesis is poorly understood and BS is considered a multifactorial disease involving interactions of several genes such as the human leucocyte antigen (HLA)-B51, with unclear environmental exposures.
In line with such heterogeneity and uncertainties, the diagnosis of BS can be challenging. This narrative review focusses on clinical features of BS, highlighting their specific value on the diagnosis of the disease.
METHODS
A PubMed search was conducted, using the keywords ‘Behçet’ AND ‘manifestations’, ‘criteria’, ‘diagnosis’, ‘pathology’, ‘statistics’, ‘skin’, ‘eye’, ‘pulmonary’, ‘neurological’, ‘gastrointestinal’, ‘vascular’, ‘phenotypes’, ‘pathergy test’, and ‘HLA’. Original research articles (retrospective and prospective studies), systematic reviews, meta-analysis, and narrative reviews on the relevant topic published in the English language up to January 31st 2021 were included. A narrative review was performed giving priority to more recent, widely cited publications, as well as international guidelines, guided by the authors’ experience.
EPIDEMIOLOGY
BS has its higher prevalence in the Mediterranean region, Middle East, and Far East Asia. Because of this peculiarity, it has been referred as the ‘Silk Road disease’. In a meta-analysis by Maldini et al.,4 a pooled prevalence was reported to be 10.3/100,000 globally, 119/100,000 for Turkey, 31.8/100,000 for the Middle East, 4.5/100,000 for Asia, 3.8/100,000 for North America, and 3.3/100,000 for Europe.4 The prevalence of distinctive organ involvements also varies widely according to geographical regions and ethnic background (Table 1),5-15 as well as according to sex and age.
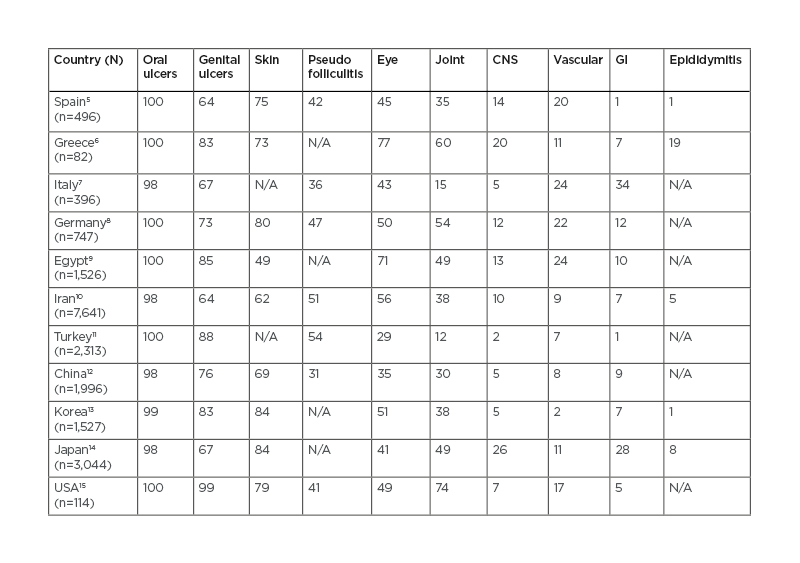
Table 1: Clinical manifestations of patients with Behçet’s syndrome from diverse world regions.
CNS: central nervous system; GI: gastrointestinal; N/A: not applicable.
Data is presented as rounded percentages.
GI involvement, for example, is reported in 1–2% of cases in some Mediterranean regions, whereas frequencies around 30–40% are reported in Far East and in patients with northern European background.10 BS most often develops in the third or fourth decade of life, but onset in childhood has been reported in 6–24% of patients.16 Compared with adults, children have more frequent neurologic manifestations, particularly cerebral venous sinus thrombosis (CVST), GI involvement and family history of BS, and less frequent ocular manifestations.17 The frequency of BS does not differ based on sex but there is difference in disease expression between sexes. Skin, eye, central nervous system (CNS), and vascular involvement are more common in males, whereas erythema nodosum (EN) and genital ulcers (GU) are more frequent in females.18 The mortality rate is highest early after disease onset and significantly increased among younger men (<25 years of age).19
CLINICAL MANIFESTATIONS
Mucosa and Skin
Mucocutaneous lesions are the most common and usually the earliest manifestations. They include oral and genital ulcerations, papulopustular skin lesions, and nodular-like lesions.20 Oral ulcers (OU) are generally the first and most common symptom,2 occurring in almost all patients. They are usually small, painful, round, or oval erosions, mainly localised on the lips, buccal mucosa, tongue, and soft palate (Figure 1A). OU typically heal spontaneously in a few days to 2 weeks and without a scar.21 Fatigue, stress, histamine-rich or -liberating food, local trauma, menstruation, and being a non-smoker have been reported as contributing factors for OU activity in BS.22,23 They continue to develop for many years after disease onset, although the frequency of the episodes tends to diminish over time.19
GU are less frequent than OU but have a high discriminatory value on the diagnosis of BS.2 They are usually located in the scrotum or in the major and minor labia (Figure 1B). More rare locations include the penis, vagina, cervix, inguinal, perineal, and perianal areas.24 GU are painful at the initial phase, beginning as papules, pustules, or necrosis. They are usually deep and heal within 10–30 days, with formation of scars.24 Unlike OU, GU tend to occur only in the early years following disease onset and disappear during the later course of the disease.19
Papulopustular lesions are also common features in BS. They are folliculitis- or acne-like lesions which appear as a papule, pustule, and comedons,25 spontaneously healing in 2–3 days without scars.20 These lesions are most commonly found on back, chest, lower limbs, and buttocks.24
EN-like lesions resemble classical EN, presenting typically with bilateral, pretibial, painful erythematous nodules. They can also be localised to the face, neck, forearms, and buttocks. They usually heal spontaneously in a few weeks, with residual hyperpigmentation.24 Histological features of these lesions are a matter of controversy, but it was reported mixed or lobular panniculitis, infiltration of variable numbers of neutrophils, lymphocytes, and histiocytes and, unlike classical EN lesions, presence of vasculitis.26,27
Musculoskeletal
Characteristic symptoms are recurrent asymmetric mono- or oligoarthritis or arthralgia, usually involving large joints, especially knees, ankles, and wrists.28 Joint involvement tends to be self-limited, typically leaving no deformity or erosion.28 Sacroiliitis seems rare in patients with BS.29
Eyes
Ocular involvement is more frequent and severe in male patients.30 It can be the first manifestation of the disease in 10–20% of the cases and typically presents during the first 2–3 years after the onset of extra-ocular signs.31
The most common eye manifestations are panuveitis (60.2%) and posterior uveitis (28.8%).30 Anterior uveitis can occur but is rarely isolated and always non-granulomatous.30 Initially, the involvement is unilateral with a remitting-relapsing course, then becomes bilateral.32
Retinal vasculitis and vitritis are the most common findings (Figure 1C).30 Hypopyon, a visible sedimentation of neutrophils in the aqueous humour, is much less common although it was once considered a hallmark of BS.30
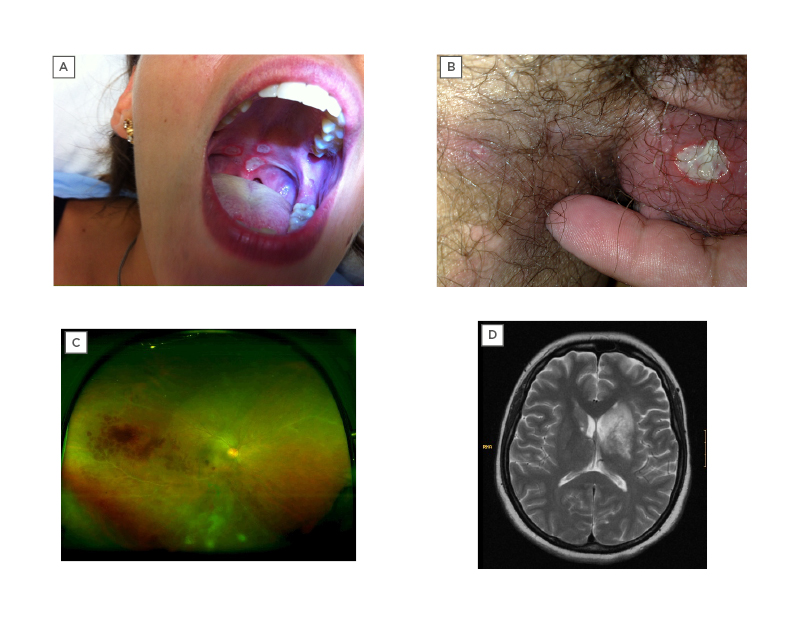
Figure 1: Examples of organ manifestations of Behçet syndrome.
A) Oral ulcers on the palate; B) genital ulcers on the scrotum; C) retinal vasculitis (ultra-widefield retinal imaging), with visible haemorrhages and contrast leakage; and D) large lesion, hyperintense on T2-weighted MRI, involving the basal ganglia and diencephalon, compatible with parenchymal neuro-Behçet syndrome.
Tugal-Tutkun et al.30 recently proposed an algorithm for the diagnosis of Behçet’s uveitis in adult patients, based on characteristic clinical findings. The items with higher accuracy for diagnosis of BS were superficial retinal infiltrate or its sequel, retinal nerve fibres layer thickness defect, signs of occlusive retinal vasculitis, diffuse retinal capillary leakage on fluorescein angiography, and absence of granulomatous anterior uveitis or choroiditis in patients with vitritis.33 Additionally, with newer diagnostic technologies such as optical coherence tomography angiography, microvascular changes in the retinal vascular plexus, and choriocapillaris have been detected before the emergence of evident clinical findings even in patients with non-ocular BS.34
Ocular involvement is a significant cause of morbidity, although the prognosis has improved in the last 20 years. Tugal-Tuktun et al.33 estimated the risk of loss of useful vision to be 6% at 1 year, 17% at 5 years, and 25% at 10 years.30
Nervous System
Neurological manifestations of BS are commonly referred as neuro-BS (NBS).35 Its frequency among patients with BS is approximately 9%, being more common in male and younger patients.36 It usually presents late in the disease course, with a mean time between onset of BS and development of NBS ranging 3–6 years.37 Rarely, it can precede other systemic features.37
The CNS is the usual site of neurological involvement.35 There are two main categories of CNS involvement: parenchymal, which is more frequent and corresponds to an inflammatory meningo-encephalitic process; and non-parenchymal, which occurs secondary to vascular involvement.35 These two subtypes occur very rarely in the same individual.38
Parenchymal-NBS usually presents with a subacute onset of brain stem, cerebral, optic, spinal cord, or diffuse syndromes, with manifestations such as pyramidal weakness, behavioural changes, headache, ophthalmoplegia, and sphincter changes.35,39 It usually follows a relapsing–remitting pattern or a primary or secondary progressive course.35 In parenchymal-NBS, cerebrospinal fluid (CSF) protein is usually modestly raised and the CSF cell count is raised in 60–80% cases, reported as neutrophilia, lymphocytosis, or mixed cellularity.35 CSF glucose is usually normal.35 MRI technique is the gold standard neuro-imaging modality for the diagnosis of NBS. A typical NBS acute or subacute lesion is large, isointense, or hypointense on T1-weighted, commonly enhanced with contrast, and hyperintense on T2-weighted, fluid-attenuated inversion recovery, and diffusion-weighted images.35,36 It is preferentially located in the brain stem, but involvement of basal ganglia, diencephalon (Figure 1D) and internal capsule is also common.36 On the other hand, chronic lesions are isointense on T1W images, smaller, and predominantly subcortical. Brain stem atrophy, particularly when asymmetric, is typical for chronic NBS and, if it is evident in the initial MRI, a progressive course could be expected.35,36 Although rare, spinal cord lesions tend to be longitudinally extensive.38
Parenchymal-NBS may be difficult to differentiate from its mimics, particularly multiple sclerosis (MS). Some differential features can be listed: sensory symptoms, optic neuritis, internuclear ophthalmoplegia, and spinal cord involvement are more common in MS. In cases of MS, chronic white matter lesions tend to be supratentorial and periventricular with corpus callosum involvement, whereas in BS, lesions are bihemispheric, subcortical, and brain stem atrophy is characteristic.2,38 In addition, CSF shows more cells in parenchymal NBS, while they are usually scarce in MS. Conversely to MS, intrathecal oligoclonal IgG bands have infrequently been reported in NBS (6.6–17.5% of patients); they are less stable in the setting of NBS compared to MS and can be easily suppressed by corticosteroid treatment.35,36
Non-parenchymal-NBS presents generally with CVST. A pseudotumour-like intracranial hypertension, acute meningeal syndrome, or acute stroke related to arterial thrombosis, dissection, or aneurysm, can also occur.35 Patients with NBS-CVST usually present with headache for a few weeks, papilledema and, occasionally, sixth nerve palsy.40 Cranial MRI scans usually show an occluded dural sinus but venous infarcts are rare.40
The prognosis of parenchymal-NBS is poor. In chronic parenchymal-NBS, a multicentre study revealed a fatal rate and bedridden state rate of 35.2% and 65.4%, respectively, at 200 months after the initiation of treatment.41 A progressive course, a higher number of attacks, relapse after steroid tapering, and elevated protein and/or pleocytosis in the CSF were associated with a poorer prognosis.42,43 CVST has, in general, a better prognosis.36
Vascular System
BS can affect both arterial and, most commonly, the venous vascular tree. Except the non-pulmonary arterial disease, vascular involvement usually presents within 5 years of disease onset.44,45 The most frequent types of vascular involvement are superficial and deep vein thrombosis, affecting most commonly the lower extremities but also the superior extremities.46 Thrombosis of atypical sites, such as inferior and superior vena cava, suprahepatic veins with Budd–Chiari syndrome, portal vein, cerebral sinuses, and right ventricle, are less frequent but quite unique to BS.20 Post-thrombotic syndrome is common. A peculiar feature of BS is the very low possibility of embolism, probably because the chronic relapsing thrombotic events transform veins into dense fibrotic structures, forming a thrombus strongly adherent to the vessel.10,44,45
Arterial involvement in BS can be occlusive or, more frequently, aneurysmal. Aneurysms can occur in peripheral and visceral arteries. Common locations are aorta, femoral, and pulmonary arteries. Presence of multiple lesions is frequent.47 The co-occurrence of venous and arterial manifestations is common in BS. The contemporary occurrence of arterial pulmonary aneurysms and peripheral venous thrombosis is the hallmark of the Hughes–Stovin syndrome, now considered a form of vascular BS.48 Screening of pulmonary artery aneurysms is important whenever anticoagulation is considered, in order to anticipate a massive haemoptysis.49
There is evidence that thrombus and aneurysm/pseudoaneurysm formation result from underlying vasculitis.45 This is the reason why the European League Against Rheumatism (EULAR) recommends immunosuppressive treatment for the management of acute thrombosis, while the use of anticoagulation is controversial.49 Although immunosuppressive treatment is effective in these manifestations, in the case of venous thrombosis, it has been described a relapse rate as high as 45% at 2 years despite treatment.50 In the case of arterial lesions, a long-term outcome study reported a complete remission in only 38.6% of patients and a relapse rate of 27.7%.47 Additionally, the vascular involvement in BS, particularly the pulmonary artery aneurysms had been identified as a leading cause of death in these patients.19
Gastrointestinal System
GI manifestations usually start 5–10 years after the onset of OU. The ileocecal area is the most commonly involved location (in up to 96% of cases), although the whole GI tract could be affected.51 It typically presents with single or few, oval or round, large, and deep ulcers, with distinct borders, usually in a focal or multifocal distribution.51 The most common symptoms include abdominal pain (87–92% of patients), followed by diarrhoea, and GI bleeding.51 Acute abdomen findings such as perforation or GI bleeding were reported in a percentage as high as 30% of patients with GI involvement from BS.52 Histologically, Behçet’s ulcers contain non-specific chronic inflammation, and, eventually, a phlebitis.52,53
GI involvement in BS may be difficult to differentiate from Crohn’s Disease. A comparative study revealed that round shape of the ulcers, five or fewer in number, focal distribution, and absence of aphthous or cobblestone lesions were significantly dominant features in BS.54 Long-segment, intestinal obstruction, fistulas, and perianal abscesses were found to be significantly more frequent in Crohn’s Disease.55 GI involvement tends to have a recurrent course. Even with treatment, relapses may occur in approximately 20% of patients.52
Cardiac Manifestations
Cardiac involvement is very rare, described in 0.6% of patients in a large cohort in Iran.10 Virtually all cardiac structures can be affected. There have been reports of coronary artery disease (with documented aneurysms and stenotic lesions, thrombus, or external compression by an aneurysm of a sinus of Valsalva), pericarditis, myocarditis, endocarditis and endomyocardial fibrosis, aortic regurgitation (caused by annular dilation and sinus of Valsalva aneurysm secondary to aortitis), mitral valve prolapse, and intracardiac thrombus (particularly in the right ventricle, and prone to recurrence).56,57
Pulmonary Manifestations
Pulmonary artery involvement (PAI) has a prevalence rate of less than 5%.58 It generally consists of pulmonary aneurysms, but isolated pulmonary artery thrombosis has also been reported. Haemoptysis is the most common symptom. Cough, fever, chest pain, and dyspnoea can also occur. Pulmonary parenchymal lesions commonly accompany active PAI. They may present as nodules, consolidations, cavities, and ground-glass lesions.59 Parenchymal lesions can be confused with opportunistic infections. However, studies have supported that parenchymal lesions are part of the PAI spectrum as they decrease significantly after immunosuppressive treatment and histopathological examination of the nodular infiltrations shows bronchiolitis obliterans organising pneumonia, granuloma, or infarction.59
Miscellaneous
Unusual manifestations have been reported. Some examples include orchiepididymitis, which is reported variably in different populations and generally follows other clinical manifestations of BS;60 audio-vestibular system involvement, with sensorineural hearing loss and tinnitus;61 and laryngeal manifestations, with potentially destructive ulcerations and stenosis.62
DISEASE PHENOTYPES
The above-mentioned organ involvements rarely occur as discrete BS manifestations and are commonly clustered. A number of cluster analyses and association studies identified significant associations among specific disease manifestations, reported as BS phenotypes. Three major BS phenotypes have been described. The presence of a phenotype can alert for the diagnosis, may help define molecular mechanisms leading to the co-development of different manifestations and could in the future guide a strategy for a personalised therapeutic approach.
Mucocutaneous and Articular Phenotype
There were a consistent proportion of patients present with both mucocutaneous and articular involvements. It was identified an association between papulopustular lesions and arthritis, and a strong association between EN and GU.63 It was suggested that enthesitis was also part of this cluster. Additionally, mucocutaneous lesions of the genital area were found to be negatively associated with the presence of ocular and neurological involvements.20
Peripheral Vascular and Extraparenchymal Neurological Phenotype
In a previous study, a significant association was found between peripheral vascular disease and extraparenchymal neurological involvement.44 In addition to this, approximately 80% of the patients with PAI were found to have concomitant venous thrombosis.64 These associations suggest that events on both the arterial and the venous side of the vascular tree were likely sustained by similar pathogenic mechanisms. Vascular involvement has also been negatively associated with the presence of eye manifestations.20
Parenchymal Neurological and Ocular Phenotype
Growing evidence suggests an association between the posterior uveitis and parenchymal neurological involvement. Male sex and HLA-B51-positivity are features associated with this phenotype.65
DIAGNOSIS
Classification Criteria
There is no specific or pathognomonic biomarker, histopathology feature, or laboratory test for the diagnosis of BS; therefore, BS diagnosis is primarily clinical. Development of classification and diagnostic criteria have been an important focus in this field in order to categorise patients for study purposes and, in some cases, with the intent to guide an accurate diagnosis.
Classification and diagnosis criteria are produced using the same methodology, but they have different purposes. Classification criteria are intended to define a homogeneous population with similar clinical features, and, for that reason, they should have high specificity for the underlying disease while paying the price of losing sensitivity. Conversely, the goal of diagnostic criteria is to accurately identify as many individuals with the condition as possible, and therefore they should have high sensitivity and specificity, as well as high predictive measures. In theory, a diagnosis applies classification criteria to an individual patient but the diagnostic performance of any criteria depends on their sensitivity and specificity, and also on pretest probability of the disease, which reflects the prevalence of the disease and potential mimickers. In the case of BS, because of the wide clinical heterogeneity as well as the geographical and ethnic variation of prevalence and disease expression, it is difficult to accomplish universal diagnostic criteria that capture the full range of disease presentations and perform equally well in diverse populations.
Reflecting this challenging task, to the date of this article, 17 sets of diagnosis/classification criteria for BS have been proposed; the majority originating from different countries.66 In general, they have OU, GU, and eye involvement in common. The first international criteria were the International Study Group (ISG) criteria for Behçet disease,67 developed in 1990 with the collaboration of seven countries and being the most widely used thereafter. As a result of the low sensitivity of the ISG criteria observed in some validation studies, the International Team for the Revision of the International Criteria for Behçet disease (ITR-ICBD)68 proposed new criteria in 2014, with contributions from 27 countries. The ICBD intended to perform well regardless of the country and provide a useful tool to the identification of possible BS by non-experts. Compared to the ISG criteria, the ITR-ICBD consider different points for distinct manifestations, oral aphthae are not a mandatory criterion, and include vascular and neurological findings (Table 2). ICBD was shown to have much better sensitivity, 3% lower specificity, and better accuracy than ISG criteria.66 The ICBD were then validated in Iranian patients69 but its performance on a British cohort revealed a specificity of only 19%,70 again advising caution on its use for diagnosis purposes in diverse clinical sceneries. Therefore, criteria have to be tailored to the practice setting and applied with clinical judgment. The pursuit of universal diagnostic criteria is now debatable, and subspecialty specific criteria have been proposed as a way to reduce the number of differential diagnosis in each particular scenery.71
Based on a childhood registry, a classification criteria for paediatric disease has been developed, called Paediatric Behçet’s Disease criteria (Table 2).72 In these criteria, all manifestations have the same weight, oral aphthosis is not mandatory, and a pathergy test is not considered.
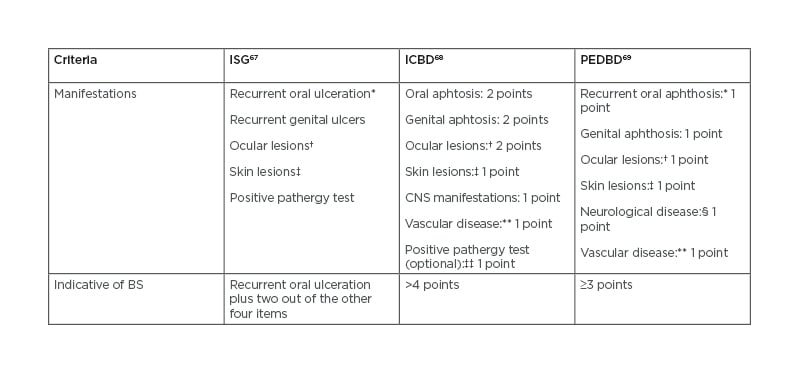
Table 2: Comparison of the International Study Group, International Criteria for Behçet disease, and Paediatric Behçet’s Disease criteria.
*Oral ulceration needs to be recurrent and ≥3 in 12-month period for ISG and PEDBD.
†Defined as anterior and/or posterior uveitis and/or retinal vasculitis.
‡In ISG as erythema nodosum-like, pseudofolliculitis, papulopustular lesions, or acneiform nodules; in ICBD defined as erythema nodosum, pseudofolliculitis (pustulosis), or aphthous ulcers; in PEDBD as erythema nodosum, necrotic folliculitis, acneiform lesions.
§Excludes isolated headaches.
**In ICBD defined as arterial thrombosis, large vein thrombosis, phlebitis, and superficial phlebitis; in PEDBD as venous thrombosis, arterial thrombosis, arterial aneurysm.
††If pathergy testing is conducted, one extra point may be assigned for a positive result.
BS: Beçhet’s syndrome; CNS: central nervous system; ICBD: International Criteria for Behçet disease; ISG: International Study Group; PEDBD: Paediatric Behçet’s Disease.
Pathergy Test
The pathergy test is a non-specific hypersensitivity skin reaction induced by trauma such as a needle prick. Although there is no standardised procedure, it generally consists of an intradermal puncture on the skin with a 20-gauge or smaller needle into the patient’s flexor aspect of the forearm.
It is considered positive when an indurated papule or pustule forms within 48 hours. Pathergy positivity is highly suggestive but not pathognomonic of BS, occurring for example in pyoderma gangrenosum, Sweet syndrome, and inflammatory bowel diseases.73
There are significant variations in the prevalence of pathergy among different populations. The positivity rate in BS is highest in countries along the Silk Road, and it is uncommon in Northern European and North American patients with BS.74 Additionally, numerous studies have also demonstrated a decline in the prevalence of positive pathergy tests over the past decades.10 Taking this into account, pathergy testing may be considered especially for patients who do not fulfil the criteria, standing near to the cut-off point, at least in countries with high prevalence of BS.
HLA-B51
BS is associated with the major histocompatibility complex HLA-B51 allele. HLA-B51 is carried by 34% to 64% of patients and increases the risk of BS development by a factor of 5.9.75 Nevertheless, its prevalence varies across the globe, being higher in Asian, Middle Eastern, and Southern European populations, and lower in Northern Europe and North America.75 Additionally, the presence of HLA-B51 genotype among control healthy individuals ranges from 11% to 22%.75 Although HLAB51 allele is the most established risk factor for BS, it is neither necessary nor sufficient for its development. For these reasons, the diagnostic value of HLA-B51 positivity is limited and it may be used as a supportive finding only in the presence of appropriate clinical findings.
The presence of HLA-B51 seems to have some relation to disease expression. According to a meta-analysis, HLA-B51/B5 is associated with significant increased prevalence of GU and ocular or skin involvement and with lower risk of GI involvement in BS.76 Conversely, a recent large Japanese survey found that the presence of HLA-B51 correlated negatively with the presence of GU.14 In any case, clinical presentations of HLA-B51-positive and negative BS patients are not distinguishable.
CONCLUSION
BS is a complex and heterogeneous entity that does not fit perfectly into any recognised nosological group. OU are the most frequent manifestation, but the vascular and neurological involvements have the worst prognosis. Diagnosis is not straightforward because it is mainly based on clinical features. There are several classification criteria, often used to assist the diagnosis, but limitations should be considered. A solid knowledge of the typical manifestations and the local presentation of the disease is therefore essential to an accurate diagnosis. Current research focusses on the pathogenic mechanisms of the disease and its phenotypes, as well as on advances in laboratory and imaging techniques for diagnosis and monitorisation of BS. Shedding light on these topics may contribute to tailored therapeutic strategies and better outcomes in the future.