Meeting Summary
Two symposia occurred during the European Respiratory Society (ERS) Congress 2024, highlighting the heterogeneity in chronic inflammatory pathways that underlie chronic obstructive pulmonary disease (COPD) pathophysiology. In ‘A Breath of Fresh Air: A Greater Understanding of COPD With Type 2 Inflammation’, Henrik Watz (Chair), German Center for Lung Research, Grosshansdorf, Germany, provided an overview of both the pathophysiology and the burden of disease of COPD. He discussed how exacerbations, which may be increased in those with evidence of Type 2 inflammation, contribute to the cycle of worsening COPD. Mona Bafadhel, King’s College London, UK, provided an examination of the mechanisms and biomarkers of Type 2 inflammation in COPD. Finally, Alberto Papi, University of Ferrara, Italy, summarised the latest research on biological treatments targeting Type 2 inflammation in COPD. The second symposium, ‘Targeting interleukin-33 (IL-33) in COPD: Exploring New Frontiers for COPD Management’, discussed inflammation in COPD, focusing on the central role of IL-33 as a mediator for both Type 2 and Type 1/Type 3 inflammation. Klaus Rabe (Chair), LungenClinic, Grosshansdorf, Germany, reviewed the structure and function of IL-33 and its initial processes that lead to downstream immune responses. Stephanie Christenson, University of California, San Francisco, USA, explored how genetic and environmental factors contribute to IL-33 activity in COPD pathology. Next, Paola Rogliani, University of Rome ‘Tor Vergata’, Italy, presented an examination of IL-33 inflammatory processes and evidence from COPD animal models illustrating the role of IL-33 in airway inflammation and lung function decline. Klaus Rabe concluded with an examination of IL-33 as a target for new COPD treatment approaches.
Introduction
A persistent, often progressive, lung disease, COPD results from chronic inflammation that leads to structural changes resulting in airway remodelling and airflow obstruction.1,2 However, just as individuals with COPD can present with different symptom patterns, the inflammatory pathways involved in COPD pathophysiology are also heterogeneous.1 Although Type 1/Type 3 inflammation drives COPD in most individuals, up to ~30−40% have Type 2 inflammation, and there is also potential overlap in inflammatory mechanisms.3-5
A Breath of Fresh Air: A Greater Understanding of COPD with Type 2 Inflammation
Living with COPD: Linking Inflammation with Patient Burden
Henrik Watz
Chronic inflammation, triggered by irritants and oxidative stress (eg., tobacco smoke, pollutants), is the key factor driving the airway and parenchymal structural changes that characterise COPD and limit airflow in the lungs (Figure 16).1,2,7 Pathophysiological changes in the airway specifically linked to chronic inflammation in COPD include barrier disruption, goblet cell hyperplasia, inflammation and excess mucus, and remodelling. Parenchymal structural changes include breakdown of the alveolar membranes (emphysema) and air trapping.1,2 Smoking, pollutants, environmental or occupational exposure, abnormal lung growth and development, and genetics and early life events all increase the risk of chronic inflammation leading to COPD.1 Even in individuals with COPD who have stopped smoking, the risk of exacerbations, morbidity, and mortality remains elevated for years.8
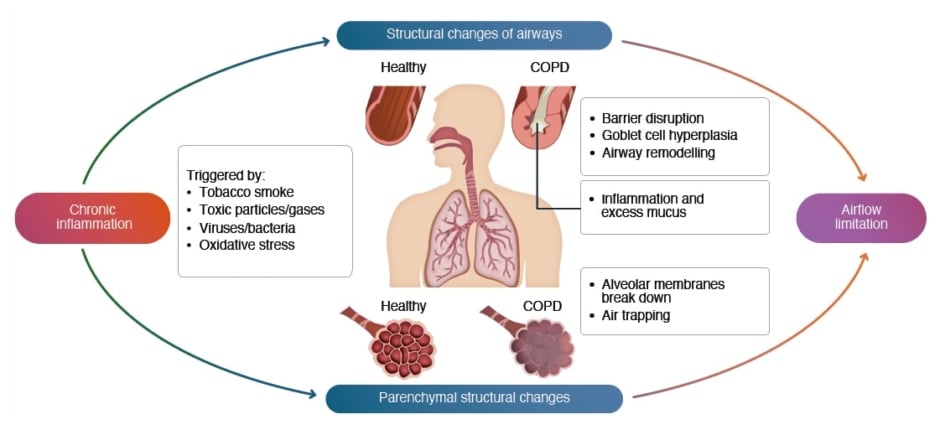
Figure 1: COPD is driven by chronic inflammation and structural changes.6
COPD: chronic obstructive pulmonary disease.
Traditionally, COPD was informally categorised based on the clinical observation of predominant symptoms, including the ‘pink puffer’ emphysema phenotype, associated with dyspnoea, hyperinflation, air trapping, and cachexia, and the ‘blue bloater’ chronic bronchitis phenotype, associated with chronic productive cough, respiratory infections, exacerbations, and being overweight.1,9 These characterisations were of limited clinical utility, only applicable for describing severe disease, and insufficient to encompass COPD pathophysiological or phenotype heterogeneity.9,10 Currently, COPD categorisation reflects an evolving, broader disease spectrum that includes emphysema- and bronchitis-predominant phenotypes, as well as a frequent exacerbator or rapid decliner phenotype, and two endotypes based on the underlying inflammatory pathway: 1) Type 1/Type 3 and 2) Type 2.1,9,11-14
In addition to increasing morbidity and mortality, COPD places a heavy physical and emotional burden on individuals and contributes to physical debilitation and poor health-related quality of life.1,15,16 In interviews, individuals with persistent COPD symptoms cite exhaustion, breathlessness, feelings of depression and dread, limits on physical activity, and difficulties in lying down at night to sleep.17
More than 80% of individuals with COPD are estimated to have >1 comorbid condition.18 Associated pulmonary conditions include bronchiectasis, asthma, pulmonary arterial hypertension, and an increased risk of lung cancer.1,19,20 Extrapulmonary conditions seen in individuals with COPD include cardiovascular (CV) conditions, such as heart failure, ischaemic heart disease, and arrhythmias; osteoporosis; renal failure; metabolic syndrome; diabetes; gastro-oesophageal reflux disease; and neurologic or psychiatric conditions.1,19-21 Multi-morbidity significantly impacts quality of life and health outcomes and is associated with increased COPD exacerbations, hospitalisations, medical costs, and mortality.22
Exacerbations of COPD increase disease burden, hospitalisations, and re-hospitalisations, and are associated with a poor prognosis. An analysis of 32 studies found 9−26% and 18−39% of COPD hospitalisations resulted in readmission within 30 days and 90 days, respectively. The most common significant risk factors for readmission were comorbidities, previous exacerbations and hospitalisations, and longer initial hospital stay, with frequent COPD exacerbations increasing readmission risk 2.5-fold.23 Patients who experience more severe exacerbations tend to have an increased incidence of CV events.24 Exacerbations also lead to an increased risk of CV death, whether the patient is hospitalised or not.25 Increased frequency and severity of exacerbations have been associated with increased rates of all-cause mortality, COPD-related mortality rate, and future exacerbations.26,27 Importantly, repeated exacerbations lead to progressive and potentially irreversible lung damage. Following a moderate or severe COPD exacerbation, forced expiratory volume in 1 second (FEV1) may not reach a patient’s pre-exacerbation levels, even after ≥1 month.28 Further, individuals who have frequent exacerbations show a significantly faster decline in FEV1 than those with infrequent exacerbations.29 Severe lung function impairment has been shown to be significantly associated with increased 90-day post-discharge mortality.30 In all, low FEV1 is a risk factor for COPD exacerbations and hospitalisations, contributing to a vicious cycle of exacerbations and declining lung function.13
Evidence from studies investigating increased blood eosinophil (EOS) counts, which can correspond to increased lung EOS numbers and elevated markers of Type 2 inflammation in the airways, supports a higher disease burden for individuals with COPD associated with Type 2 inflammation.1,31 An analysis of data from the Genetic Epidemiology of COPD study in 1,553 individuals with COPD found the risk for COPD exacerbations increased with increasing blood EOS counts, particularly for those with blood EOS ≥300 cells/µL.31 The Canadian Cohort of Obstructive Lung Disease study examined blood EOS counts in participants aged ≥40 years with and without COPD. The study found that the annual decline in FEV1 for those with EOS ≥300 cells/µL was significantly greater than for those with lower EOS levels (P≤0.01).32 Elevated EOS count (≥300 cells/µL) has been shown to be a marker for COPD-related rehospitalisation at 30 days up to 12 months and for all-cause rehospitalisation at 90 days up to 12 months.33 In an observational study, individuals with COPD with ≥300 EOS cells/µL were 3.21 times more likely to be readmitted to the hospital for COPD than those with EOS <300 EOS cells/µL.34
In summary, Watz emphasised that worsening disease in COPD contributes to a vicious cycle.35 Increased chronic inflammation and its attendant structural damage worsens symptoms and increases exacerbation risk, leading to a progressive decline in lung function.1,2,7
Unravelling the Role of Type 2 Inflammation in COPD
Mona Bafadhel
The Type 1/Type 3 and Type 2 endotypes of COPD have distinct characteristics.1,14 Type 1/Type 3 inflammation is marked by elevations in sputum neutrophils, Type 1 cytotoxic and T helper (Th) 1 and 17 cells, and innate lymphoid cell (ILC) 3, whereas Type 2 inflammation markers include elevated sputum EOS, Th2 cells, ILC2, and Type 2 cytokines IL-4, IL-13, and IL-5.36-40
Bafadhel underscored the potential roles of Type 2 cytokines in COPD with Type 2 inflammation, including airway remodelling, goblet cell hyperplasia, mucus production, and EOS trafficking to tissues (Figure 26).41-45
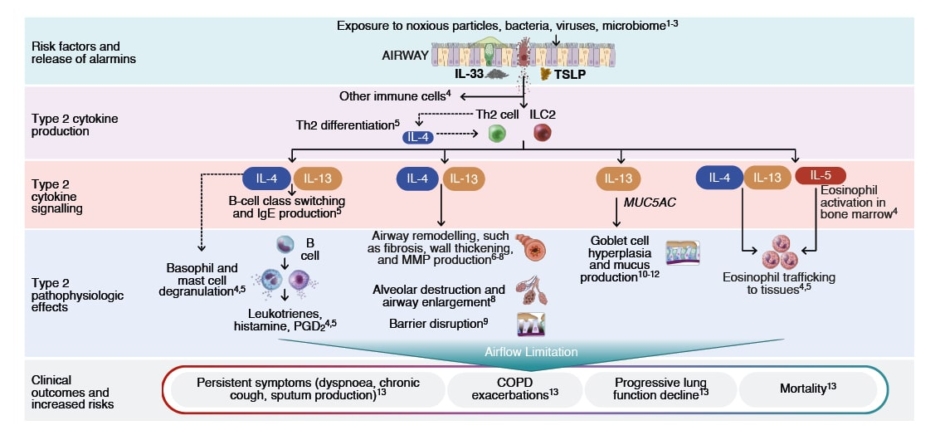
Figure 2: Potential roles of Type 2 cytokines in COPD with Type 2 inflammation.6
COPD: chronic obstructive pulmonary disease; IgE: immunoglobulin E; IL: interleukin; ILC2: group 2 innate lymphoid cell; MMP: matrix metalloproteinase; MUC: mucin; PGD2: prostaglandin D2 ; Th2: T helper cell Type 2; TSLP: thymic stromal lymphopoietin.
Blood EOS is recognised by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) as a biomarker for inhaled corticosteroid (ICS) use, with levels ≥300 cells/μL indicating a higher likelihood that ICS may prevent COPD exacerbations.1 However, COPD endotyping remains an emerging concept, and the lack of established biomarkers to differentiate inflammatory profiles complicates the treatment decision-making process.11,46,47 To date, fibrinogen remains the only circulating biomarker approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) to assess the risk of COPD exacerbations and mortality.48 Other biomarkers that have been investigated in relation to COPD clinical outcomes include blood and sputum EOS count, fractional exhaled nitric oxide (FeNO), and alpha-1 antitrypsin.10,48,49
Bafadhel highlighted the potential of biomarkers to improve COPD understanding and treatment. She emphasised their practicality and ease of use in clinical settings, suggesting that with further research, these biomarkers could play a larger role in personalised COPD therapy. To highlight these points, she summarised several studies illustrating the potential utility of EOS and FeNO in assessing COPD disease and treatment. In a study of 226 individuals with COPD, the annualised rate of moderate and/or severe acute COPD exacerbations was significantly higher in individuals with ≥2 visits where FeNO measured ≥20 parts per billion than in individuals with 0 or 1 visit (P<0.05).50 Elevated EOS was associated with an increased incidence rate ratio of severe and moderate COPD exacerbations in a study of >7,000 individuals with COPD participating in the Copenhagen General Population Study.51 Another analysis of data from this study found that blood EOS ≥300 cells/µL and FeNO ≥20 parts per billion separately and together were significantly associated with FEV1 decline (P≤0.004).52 Lastly, Bafadhel showed data supporting EOS and FeNO as biomarkers to predict ICS responsiveness in COPD. In a post hoc analysis of a randomised trial, annual COPD exacerbation rates were lower with budesonide+formoterol than with formoterol alone in individuals with blood EOS ≥340 cells/μL, but not in individuals with blood EOS <100 cells/μL.53 A separate study showed that ICS plus a long-acting β2 agonist reduced COPD Assessment Test scores by 7.20 points from baseline in individuals with COPD and high baseline FeNO, but was not as effective for improving this measure in those with low baseline FeNO.54
On Target: Perspectives From the Evolving Biologic Landscape in COPD
Alberto Papi
The 2024 GOLD algorithm for therapy escalation after a COPD exacerbation uses blood EOS levels to guide decisions.1 According to GOLD, all individuals experiencing an exacerbation should be treated with a long-acting β-agonist plus a long-acting muscarinic antagonist, but those with EOS ≥300 cells/μL also should receive ICS.1 Despite the optimisation of inhaled treatment with triple therapy, some individuals remain at risk for exacerbations. Papi reviewed the evolving COPD treatment landscape and presented data from clinical studies in COPD for biological therapies targeting steps in the Type 2 inflammatory pathway.
Targeting IL-33 in COPD: Exploring New Frontiers for COPD Management
Understanding the Role of IL-33 Size and Activity in the Pathophysiology of COPD
Klaus Rabe
Rabe reviewed the complexity and diversity of the inflammatory pathways associated with COPD pathology and discussed the structure and function of IL-33. Full-length IL-33 (IL-33FL) secretion from airway epithelial cells is induced in response to inhaled irritants, pathogens, and allergens. In this form, IL-33FL can be cleaved by proteases from inflammatory cells or allergens to form hyperactive, mature IL-33, which has 30- to 60-fold higher activity than IL-33FL, or inactivated by oxidation, sequestration, and proteolytic degradation. Mature, activated IL-33 stimulates ST2+ immune cells, triggering both Type 1 and Type 2 inflammation that can underpin COPD pathology. However, inactivated IL-33 may induce ST2-independent signalling and also promote a COPD phenotype.55,56 Because of its key role in the pathophysiology of different COPD endotypes and phenotypes, IL-33 is a promising therapeutic target.
What Do We Know About IL-33 Genetics?
Stephanie Christenson
Christenson discussed how IL-33 activity in COPD pathology involves a convergence of genetic variations and environmental exposures. To characterise the impact of IL-33 genetics on COPD risk, investigators performed an analysis of IL-33 loss-of-function (LOF) and gain-of-function (GOF) variants known to affect asthma risk among individuals of European ancestry utilising data from the UK Biobank and Geisinger Health System studies in COPD.57 Three variants were selected for analysis: 1) a rare IL33 LOF slice-acceptor allele that reduces total IL33 messenger RNA (mRNA) and decreases asthma risk; 2) a common IL33 intronic GOF variant that increases IL33 mRNA and asthma risk; and 3) a common IL1RL1 (gene that encodes the IL-33 receptor) intronic GOF variant that lowers plasma levels of soluble IL-33 receptor and increases asthma risk. The results showed that the rare LOF variant was associated with a 21% reduction in the risk of COPD and a reduction in blood EOS (P<0.0001), but the two common GOF variants were associated with significantly increased odds of COPD (P<0.05 for each variant).57
Next, Christenson reviewed how smoking influences IL-33 activity in COPD and highlighted the importance of considering the complex effects of smoking on IL-33 activity within the context of COPD. An analysis of IL-33 levels in 62 individuals with COPD showed that smoking pack-years was positively correlated with IL-33 expression in the lungs (R=-0.58). In this study, a larger proportion of individuals with high IL-33 had previously regularly smoked cigarettes than patients with low IL-33; in contrast, those who were currently regularly smoking cigarettes tended to have low IL-33.58 This trend of individuals who currently regularly smoke having lower IL-33 expression than those who formerly regularly smoked has been confirmed in multiple studies of samples collected by bronchial biopsy or bronchial brushings.59 This finding may be due to the shifting of basal cells towards a more differentiated state in those who currently regularly smoke, and may explain the clinical observation of greater efficacy of anti-IL-33 treatment in those with COPD who previously regularly smoked.59 However, even in those with COPD who currently regularly smoke, high IL-33 expression has been associated with lower percent predicted median residual volume and lower percent predicted and percent mean residual volume to total lung capacity.60
The Role of IL-33 in COPD Pathophysiology and Its Impact on Disease Remodelling
Paola Rogliani
Rogliani began with a closer look at the key role IL-33 plays in COPD as an initiator and amplifier of broad inflammatory cascades. Stimulation from smoke, pollutants, infections, or oxidative stress induces IL-33 production and secretion from the airway epithelium and the endothelium of blood vessels in the lungs.55,61 IL-33 then activates immune cells and non-haematopoietic lung cells to promote immune responses and increase inflammation (Figure 362).61 As a central mediator of inflammation in COPD, IL-33 drives both Type 1/Type 3 and Type 2 inflammatory responses, and stimulation of IL-33 can lead to increased neutrophil and blood EOS levels.5,45,63 The chronic inflammation initiated and amplified by IL-33 results in airway remodelling, including airway smooth muscle thickening and fibrosis in small airways.7,64
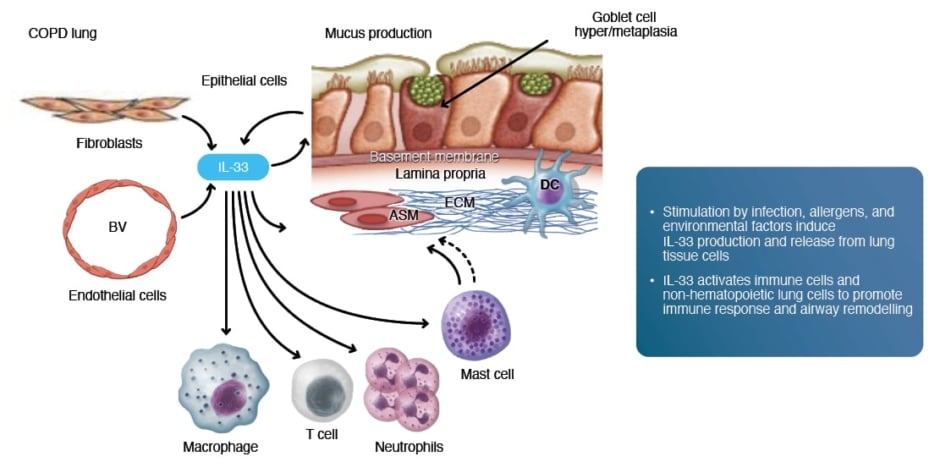
Figure 3: IL-33 mediation of immune response and airway obstruction and remodelling in COPD.62
ASM: airway smooth muscle; BV: blood vessel; COPD: chronic obstructive pulmonary disease; DC: dendritic cell;
ECM: extracellular matrix.
Evidence from animal models illustrates how IL-33 plays a role in both Type 1/Type 3 and Type 2 inflammatory responses.65,66 In mice, systemic IL-33 overexpression produced through hydrodynamic DNA delivery induced significant neutrophilia, associated with Type 1/Type 3 inflammation, and eosinophilia, associated with Type 2 inflammation, in lung tissue, as well as production of Type 1 and Type 2 inflammatory cytokines.65 A separate study in mice found that prolonged exposure to house dust mites resulting in upregulation of IL-33 induced features of severe airway diseases, such as smooth muscle thickening, increased collagen deposition, and epithelial degeneration.66 Although initial inflammatory responses had Type 2 characteristics, including increased EOS, longer exposures led to a mixed inflammatory phenotype, with increased neutrophils and other markers of Type 1/Type 3 inflammation.66
IL-33: A Promising Target
Klaus Rabe
Rabe reviewed the landscape of available therapies and those in development that target the chronic inflammation underlying COPD pathophysiology. He also presented data supporting IL-33 as a potential target for COPD treatment. Ongoing research is refining the understanding of IL-33 in COPD, focusing on identifying ideal patient subgroups for IL–33–targeted therapies, and evaluating the long-term efficacy and safety of these interventions. In all, Rabe articulated a shared vision of more personalised approaches in medicine, where treatments are tailored to individual patients based on their unique biological mechanisms.
Conclusion
As illustrated by the presentations in these symposia, COPD is a heterogeneous condition with variations in symptomatology, the events that trigger the chronic inflammation driving the disease processes, and the nature of the inflammatory pathways. Understanding the heterogeneity of inflammation in COPD pathophysiology helps to guide diagnosis and treatment. Optimal COPD treatment is critical to arresting the vicious disease cycle of exacerbations and worsening lung function that increases disease morbidity and mortality.
MAT-GLB-2406080 – 1.0 – 10/2024