![]()
Corrigendum: Modulating the Expression of Multiple Surface Receptors on Epithelial Cells and Promoting Lung Macrophage Anti-viral Functions by OM-85 Inhibits Severe Acute Respiratory Syndrome Coronavirus 2 Infection
Authors: Niki Ubags, Christophe von Garnier
Original citation: EMJ Respir. 2022;10[1]:67-76.
Date correction published: 22.12.21
In the article in EMJ Respiratory 10[1] 2022 (pages 69–70) Figures 1 and 2 (D–F) did not include relevant references and copyright statements. This has now been corrected; Figure 1 was adapted from Zhang et al.,17 Figure 2D was adapted from 1 image by Zipeto et al.18 (2020), Figure 2E was adapted from 1 image by Kinaneh et al.19 (2021) under the terms of the Creative Commons Attribution License (CC BY), and Figure 2F was reproduced with permission from Garantziotis and Savani (2019), who retain its copyright.
EMJ apologises for the error and any inconvenience caused.
![]()
Summary
The emergence of a new virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in Wuhan, China, in December 2019 triggered a global pandemic, forcing much of the world to adopt lockdown strategies and leading to extraordinary threats to the global healthcare system. The clinical manifestations of the disease, referred to as COVID-19, range from mild, self-limiting flu-like respiratory illness to life-threatening multi-organ failure and death. The rapid progress in our understanding of COVID-19 pathogenesis has led the development of effective vaccines, monoclonal antibodies, and anti-viral agents. However, a major cause of concern is the continuous and rapid emergence of new mutations that can progressively decrease sensitivity to the existing anti-COVID-19 tools. Safe, affordable, and widely available treatments are therefore urgently needed to reduce the frequency and/or severity of SARS-CoV-2 infection. OM-85 is a standardised lysate of bacterial strains widely used for the prophylaxis of airway recurrent infections in adults and children with an excellent safety profile. In experimental animal models and in clinical trials this compound was shown to possess anti-viral activities through immunomodulatory responses, but also by inhibiting infection. The positive results reported in models of common respiratory virus infection has recently encouraged researchers from three independent groups to evaluate whether OM-85 could also affect SARS-CoV-2 infection. The results of these studies are summarised in this review.INTRODUCTION
SARS-CoV-2, a member of the coronavirus family, was first detected in December 2019 in Wuhan, China, and identified as the causal agent of a viral pneumonia named COVID-19.1 COVID-19 rapidly became pandemic, and has led to an extraordinary threat to the global healthcare system and economic stability. SARS-CoV-2 infection is characterised by a wide clinical spectrum, ranging from asymptomatic infection to a multi-organ dysfunction disorder leading to respiratory failure and death.2,3 The severity of the disease is related to a variety of factors that, in addition to SARS-CoV-2 virulence, involve a deficient innate and adaptive immune response associated with an exaggerated inflammatory reaction.3 The rapid development and global distribution of effective vaccines, monoclonal antibodies and anti-viral agents have greatly improved control of the infection and its related health complications.4 However, the continuous, fast emergence of new mutations can significantly reduce the protection elicited by the existing anti-COVID-19 prevention and treatment tools.5,6 Novel, relatively inexpensive, safe, and widely available preventative and therapeutic treatment strategies which inhibit viral infection and replication and stimulate the host immune response are therefore urgently needed. OM-85, an oral standardised bacterial lysate isolated from human respiratory strains, possesses strong immunomodulatory properties, and is widely used to prevent recurrent infections and/or exacerbations in at risk populations. Specifically, it has been shown to exhibit anti-viral activities in cell culture experiments, experimental animal models, and in clinical trials.7-12 These observations prompted researchers from three independent groups to evaluate whether OM-85 could also affect SARS-CoV-2 infection.13-15 The results of these studies are summarised and discussed in this review.
OM-85 DOWNREGULATED SARS-COV-2 ACE2 RECEPTOR AND TMPRSS2 TRANSCRIPTION AND EXPRESSION
SARS-CoV-2 infection targets airway epithelial cells through the structural spike (S) protein, which contains a receptor-binding domain specifically recognising the angiotensin-converting enzyme 2 (ACE2) protein as its receptor on host cells (Figure 1A).2 After initial binding of the ACE2 receptor, proteolytic cleavage between the S1 and S2 subunits by the transmembrane serine protease 2 (TMPRSS2) is required to facilitate fusion of the viral and the host cellular membranes and for viral entry (Figure 1B).2 Experimental studies using murine models, cells derived from SARS-CoV-2 target organs, and human bronchial cells, have demonstrated that exposure to OM-85 can effectively inhibit the transcription of ACE2 and TMPRSS2 proteins.13
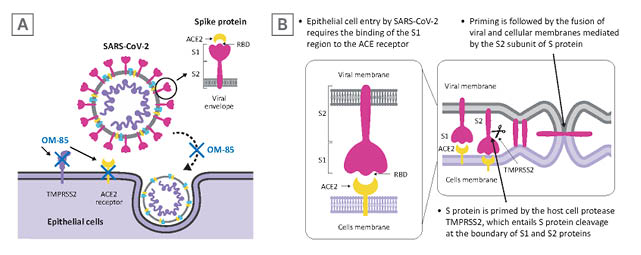
Figure 1: SARS-CoV-2 infection of epithelial cells.
A) SARS-CoV-2 targets airway epithelial cells through the structural S protein, which contains a receptor-binding domain specifically recognising the ACE2 protein as its receptor on host cells. B) Binding of the S1 subunit to ACE2 receptor and virus attachment to the host cell is followed by TMPRSS2-induced cleavage of the S2 subunit from S1 subunit necessary for virus-host cell membrane fusion.
Adapted from Zhang et al.17
ACE2: angiotensin-converting enzyme; S: spike; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; RBD: receptor binding domain; TMPRSS2: transmembrane serine protease 2.
Inhibition of Angiotensin-Converting Enzyme 2 and Transmembrane Serine Protease 2 Transcription and Expression in Murine Lungs and Human Epithelial Cell Cultures
To further determine whether OM-85 treatment could protect non-human primate and human epithelial cells against SARS-CoV-2 infection, Pivniouk et al.13 measured ACE2 and TMPRSS2 transcription lungs of mice treated with intranasal OM-85. They observed an inhibition of ACE2 and TMPRSS2 transcription at Day 2 and Day 7 after the last OM-85 treatment.13 Given the ability of OM-85 to modulate dendritic cell functions, which are engaged in anti-viral responses and depend on myeloid differentiation primary response 88 (Myd88)/TIR-domain-containing adapter-inducing interferon-β (Trif) innate immune signalling,16 the involvement of this pathway in ACE2 and TMPRSS2 transcription was consequently assessed in wild type and in Myd88−/−Trif−/− mice. As few as four intranasal OM-85 instillations were sufficient to reduce ACE2 and TMPRSS2 transcription in lung cells isolated from wild-type mice by more than 55%, whilst negligible changes were observed in Myd88−/−Trif−/− mice (Figure 2A).13
The inhibitory activity of OM-85 was then tested on cell lines derived from three SARS-CoV-2 target organs: Vero E6 cells, non-human primate derived kidney epithelial cells; Calu-3 cells, human lung cancer cells; and Caco-2 cells, human colorectal adenocarcinoma cells (Figure 2B).13 OM-85 had a significant and strong inhibitory effect on ACE2 and TMPRSS2 transcription in Vero E6 cells after 24- and 48-hour stimulation, whereas, in Calu-3 cells, significant inhibition of ACE2 transcription was only detected after 72 and 96 hours and of TMPRSS2 transcription, at 24, 72, and 96 hours. Moreover, 48-hour stimulation with OM-85 significantly reduced ACE2 and TMPRSS2 transcription in intestinal Caco-2 cells.13 Taken together, the overall OM-85-induced downregulation of ACE2 and TMPRSS2 transcription occurred more rapidly in Vero E6 cells compared with the other two cell lines.
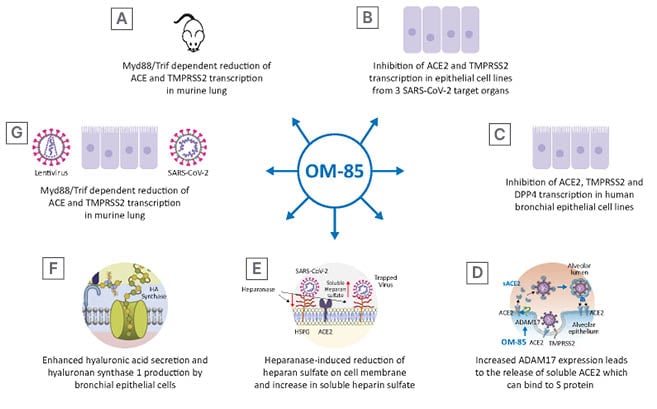
Figure 2: OM-85 can interfere with various step of the infection cycle, decreasing virus adhesion to the cell membrane and virus entry by inhibiting cell surface proteins recognised by the binding site of the S protein, promoting the release of the soluble form of some of these receptors and the synthesis of hyaluronic acid which can inhibit virus cycle at different stages.
Figure 2D was adapted from 1 image by Zipeto et al.18 (2020). Figure 2E was adapted from 1 image by Kinaneh et al.19 (2021) under the terms of the Creative Commons Attribution License (CC BY). Figure 2F was reproduced with permission from Garantziotis and Savani (2019), who retain its copyright.
ACE2: angiotensin-converting enzyme; ADAM17: a disintegrin and metalloprotease 17; DPP4: dipeptidyl peptidase-4; HA: hyaluronic acid; HPSE: heparanase; Myd88: myeloid differentiation primary response 88; S: spike; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; TMPRSS2: transmembrane serine protease 2; Trif: TIR-domain-containing adapter-inducing interferon-β.
In line with these findings, Fang et al.14 observed a significant reduction in ACE2 and TMPRSS2 transcription at 24 and 48 hours after OM-85 (1:50 dilution) stimulation in human bronchial epithelial cell lines (BEAS-2B and Nuli) and primary human bronchial epithelial cells (isolated from subjects without chronic inflammatory lung disease).14 In addition, kinetic studies on protein expression were performed in bronchial epithelial cell cultures undergoing daily OM-85 treatment over 4 days. OM-85 significantly reduced ACE2 protein expression at all time points, whereas downregulation of TMPRSS2 protein expression became significant only at 4 days.14
MODULATION OF THE TRANSCRIPTION AND EXPRESSION OF DIPEPTIDYL PEPTIDASE-4, A DISINTEGRIN, METALLOPROTEASE 17, HEPARANASE, AND HYALURONIC ACID BY OM-85
To evaluate the ability of OM-85 to modulate transcription and expression of multiple proteins involved in SARS-CoV-2 infection (dipeptidyl peptidase-4 [DPP4], a disintegrin and metalloprotease 17 [ADAM17], heparanase [HPSE], and hyaluronic acid [HA]), Fang et al.14 utilised two immortalised human bronchial epithelial cell lines (BEAS-2B and Nuli), and primary human bronchial epithelial cells.14
Modulation of Dipeptidyl Peptidase-4, A Disintegrin and Metalloprotease 17 Transcription and Expression
DPP4 is a serine exopeptidase expressed in several tissues, including the lung, kidney, liver, gut, and immune cells, which can be used as functional receptor by SARS-CoV-2.20,21 Daily treatment of human bronchial epithelial cells with OM-85 (1:50 dilution) significantly reduced the DPP4 messenger RNA (mRNA) levels at 48 hours post-exposure. Moreover, in kinetic studies it was observed that DPP4 protein levels significantly declined after 24 and 96 hours, whereas they reached control levels in between (48 and 72 hours).14 Interestingly, daily treatment of these cells with OM-85 significantly increased ADAM17 mRNA levels at 48 hours, and ADAM17 protein over a 4-day timespan.14 ADAM17 is engaged in the cleavage and release of a variety of membrane-anchored cytokines, cell adhesion molecules, receptors, ligands, and enzymes, including ACE2.22 As expected, OM-85 induced increased expression of ADAM17 protein was associated with ACE2 cleavage and a significantly increased extracellular release of ACE2 in its soluble form (sACE2), on Day 4 and 5 (Figure 2D).14 The levels of sACE2 protein in OM-85-treated cells were dose-dependent and became significant on Day 4 and 5 (1:20 and 1:50 dilution). Moreover, a positive linear correlation was detected between sACE2 and ADAM17 concentrations. Circulating sACE2 can bind to the virus effectively, acting as soluble decoy and blocking attachment to the membrane-bound receptor and virus entry;23 therefore, an OM-85 induced increase in sACE2 may be beneficial in prevention and amelioration of SARS-CoV- 2 infection.
Overexpression of Heparanase and Stimulation of Hyaluronic Acid Production
In human bronchial epithelial cell OM-85 treatment (1:50, and 1:100 dilution) induced an increase in HPSE secretion at 3 and 4 days after exposure.14 HPSE is an enzyme that acts both at the cell-surface and within the extracellular matrix to degrade polymeric heparan sulfate. This linear polysaccharide, which occurs as proteoglycan, is produced by epithelial cells.24 Expressed on the cell surface, heparan sulfate serves as a receptor for several viruses and, in the case of SARS-CoV-2 infection, facilitates the interaction between the S protein and its homing receptor ACE2.25,26 The observed increase in HPSE secretion following OM-85 exposure was associated with an increase of soluble heparan sulfate and a decrease of heparan sulfate expression on the cell membrane 3 and 4 days following treatment (Figure 2E).14 Moreover, a significant correlation was detected between soluble heparan sulfate molecules and HPSE concentration.14 HA is a non-sulfated glycosaminoglycan distributed throughout connective, epithelial, and neural tissues.27 Being one of the chief components of the extracellular matrix, HA participates in a variety of processes, including the response to bacterial, fungal, and viral infections.28 Stimulation of human primary epithelial cells with OM-85 led to enhanced synthesis and secretion of HA (Figure 2F). This observed elevation of HA following OM-85 exposure correlated with increased expression of hyaluronan synthase 1 (Has-1), but not of Has-2 and Has-3, after 24 hours of OM-85 stimulation. Has-1, Has-2 and Has-3 show differences regarding substrates, localisation within the cellular compartment, and cleavage products. In addition, their role in airway viral infections is largely unknown. However, the upregulation of Has-1 detected by Fang et al.14 suggests that OM-85 supports the de novo synthesis of long-chain hyaluronic acid, which has been shown to reduce viral infections.29,30
Taken together, it was observed that OM-85 exposure led to modulation of DPP4, ADAM17, HPSE, and HA. However, it is important to note that these effects are most likely indirect given that cellular signalling mechanisms to explain the effect of OM-85 on SARS-CoV-2 binding host proteins and glycosaminoglycans remain undetermined.
INHIBITION OF VIRUS ATTACHMENT AND EPITHELIAL CELL INFECTION
To investigate whether modulation of the expression of molecules involved in SARS-CoV-2 infection could also reduce SARS-CoV-2 S protein attachment to host cells, a recombinant histidine-tagged S1 subunit, comprising the SARS-CoV-2 receptor binding domain was used.13,31 Validation of the ability of this assay to specifically detect ACE2-mediated S1 protein cellular binding was assessed in non-transfected or stably transfected HEK293T cells with human ACE2 (ACE2/HEK293T), in addition to Vero E6 and Calu-3 cells.13 S1 binding was detected in only 5% of non-transfected HEK293T cells, whereas more than 99% of PBS-treated ACE2/HEK293T cells and a substantial proportion of Vero E6 (22±0.5%) and Calu-3 (24±1%) epithelial cells bound SARS-CoV-2 S1 protein. Moreover, a dose-dependently reduction in S1 binding was detected (Figure 2G).
Furthermore, to evaluate whether OM-85-induced inhibition of SARS-CoV-2 S1 protein attachment to epithelial cells also reduces S protein-mediated SARS-CoV-2 cell entry, replication-deficient lentiviral particles, pseudotyped with the SARS-CoV-2 S protein or with the G glycoprotein of the vesicular stomatitis viruses (VSV) were used.13,31 OM-85 pre-treatment strongly inhibited Vero E6 cell transduction by SARS-CoV-2–pseudotyped particles but not by VSV-G–pseudotyped particles, because VSV-G does not recognise ACE2 as receptors. Interestingly, HEK293T cells expressing the ACE2 protein (ACE2/HEK293T) were readily transduced by SARS-CoV-2 pseudotyped particles, but OM-85 failed to inhibit viral entry into these cells since these cells do not downregulate ACE2 on OM-85 stimulation. Therefore, the most important event characterising the OM-85–induced suppression of SARS-CoV-2 infection is the reduction of ACE2 expression. Similar results were obtained using pseudotyped lentivirus expressing the SARS-CoV-2 S-protein and human primary epithelial cell lines BEAS-2B and Nuli.14 Finally, to assess whether OM-85 treatment suppressed epithelial cell infection with live SARS-CoV-2, Vero cells were pre-treated with PBS or OM-85 and consequently incubated for 2 hours with SARS-CoV-2 (isolate USA-WA1/ 2020).13 SARS-CoV-2 infection was strongly and significantly inhibited in cultures pre-treated with OM-85, but not in PBS-pre-treated cultures, and this inhibition was evident even at the lowest OM-85 concentration. SARS-CoV-2 infection of Calu-3 cells was also significantly inhibited by pre-treatment with OM-85.
These results demonstrate that OM-85 inhibits SARS-CoV-2 infection of epithelial cells sourced from distinct tissues in an in vitro setting.
DECREASED MURINE CORONAVIRUS BURDEN RELATED TO PROMOTION OF LUNG MACROPHAGE ANTI-VIRAL FUNCTIONS
To identify cellular and molecular processes characterising the inhibitory effects of OM-85 on β-CoV infection, a murine coronavirus (MCoV) model was utilised by Salzmann et al.15 They evaluated the effects of oral OM-85 treatment on viral load, lung cell apoptosis, and lung macrophage anti-viral function promotion. In this study, the prototypical laboratory variant of MCoV (also called murine hepatitis virus [MHV]) A59 was used. Intranasal infection with MHV-A59 induces an acute, self-resolving respiratory infection, recapitulating the acute pneumonia observed in the majority of individuals infected with SARS-CoV-2.32,33 In addition, multi-organ involvement can be recapitulated using different infection routes. However, there are major differences between SARS-CoV-2 and MHV.34 The viral receptor for SARS-CoV-2 is ACE2 (with co-receptors), whereas carcinoembryonic antigen-related cell adhesion molecule 1a (without co-receptors) is the receptor for MHV.35 In addition, viral transmission of MHV occurs mainly via oral exposure and direct contact, whereas transmission of SARS-CoV-2 happens via inhalation or via the eyes, and can happen via both direct contact and contact with contaminated surfaces.32 Moreover, SARS-CoV-2 can infect different species, where MHV is limited to mice.
Reduction of Viral and Apoptotic Burden and Modulation of Interferon Signalling Activation
To determine whether OM-85-mediated immunomodulation was beneficial during MCoV infection, mice were pre-treated with OM-85, on 10 consecutive days, before viral infection and then analysed 2, 4, and 10 days after intranasal MCoV infection. A large reduction in viral load was observed at Day 4 post-infection and retained at Day 10.15 This observation was associated with enhanced viral clearance and downregulation of the apoptotic burden in lung tissue at 4- and 10-days post-infection (Figure 3A). The lack of adverse effects on lung pathology in the OM-85-treated animals indicated that pre-activation of the immune system did not magnify MCoV infection-induced injury. Gene ontology analysis of differentially expressed genes in infected control versus OM-85-treated animals revealed a strong activation of the immune response towards viral infection, and of pathways linked to innate immune cells and inflammation. Consistent with the knowledge that interferon (IFN) signalling is crucial in the early phases of viral infections, it was observed that the reduction in viral load observed at Day 4, i.e., early control of the infection, was associated with a significant downregulation of IFN-α, IFN-β, IFN-γ, and IFN-induced protein with tetratricopeptide repeats 1 (IFIT1) mRNA levels in lung tissue.
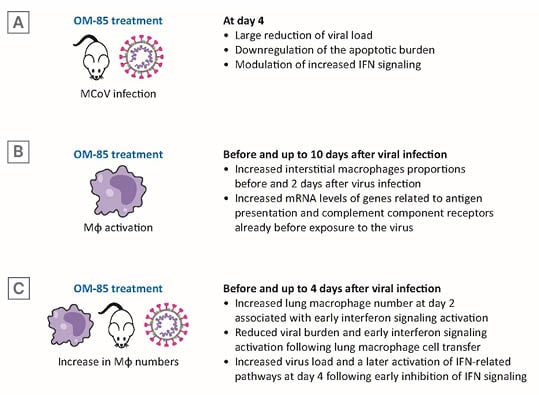
Figure 3: OM-85 treatment decreased murine coronavirus burden promoting lung macrophage anti-viral functions.
A) At Day 4, viral load and apoptotic burden reduction and modulation IFN signalling. B) Before and at Day 2, Mφs proportions and mRNA levels of genes related to anti-viral activities. C) Before and up to Day 4 after viral infection, lung Mφ number, early IFN signalling activation, effects of lung Mφ cell transfer, and of early inhibition of IFN signalling.
IFN: interferon; MCoV: murine coronavirus; mRNA: messenger RNA; Mφ: macrophage.
Lung Macrophage Expansion and Activation
RNA sequencing data indicated differences in antigen processing and presentation and macrophage activation between treated and untreated mice. Therefore, differences in lung macrophage number and state of activation were determined (Figure 3B). There are two distinct macrophage populations in the lung: alveolar macrophages, with embryonic origin; interstitial alveolar macrophages, derived from circulating monocytes;36,37 and both populations express ACE2 and TMPRSS2, required for SARS-CoV-2 attachment and are activated by SARS-CoV-2 infection.38,39 Interstitial macrophages increased after OM-85 treatment, even before virus infection, and the increase was retained 2 days post-infection. Macrophage recruitment to the lung in OM-85 treated mice was associated with phenotypical changes characterised by increased mRNA levels of genes related to antigen presentation (cluster of differentiation [CD] 86 and MHC-II) and to complement component receptors, already detectable at baseline, i.e., before exposure to MCoV infection.
To test whether the reduced MCoV load and enhanced viral clearance could be related to an early innate immune training, lung macrophage numbers and IFN pathway activation values were determined at baseline and at Day 1 and 2 after infection (Figure 3C). Increased lung macrophages numbers and INF regulatory factor 1 protein levels were detected in isolated lung cells from OM-85 treated animals. OM-85 treatment did not increase baseline IFN-α levels, but induced a strong and significant upregulation of IFN-α and of IFN-β after infection. Moreover, a strong upregulation of IFIT1 was observed with OM-85 pre-treatment from baseline (until Day 2 after virus infection). Finally, a significant OM-85-induced increase in expression was detected in the retinoic acid-inducible gene 1, an intracellular receptor involved in the identification of RNA viruses.
To assess whether early increase in lung macrophages could be sufficient to reduce MCoV infection, monocytes and macrophages isolated from naïve lung tissue were adoptively transferred to recipient mice, then infected with MCoV. Similar to the observations following OM-85 treatment, mice receiving an adoptive macrophage transfer showed a 53.91% reduction in lung viral burden compared to control mice, and reduced IFN-α, IFN-β, and IFIT1 mRNA levels, indicating a faster IFN signalling and, thereby, a faster viral resolution.
Early Blocking of Interferon Signalling Activation and Inhibition of OM-85 Anti-viral Activity
To test if the accelerated induction of IFN and its downstream targets was the main mode of action in OM-85 treated lung tissue viral defence, blocking antibodies to IFN-α/β receptor 1 (anti-IFNAR1) were used to inhibit early IFN signalling. As expected, viral load was significantly increased in animals receiving anti-IFNAR1 treatment, which also displayed a lower activation of IFN-related pathways, compared to animals receiving OM-85 treatment prior to virus infection.15 Viral burden and IFN pathway activation was comparable in OM-85 anti-IFNAR1-treated animals and in untreated control infected animals.
DISCUSSION
SARS-CoV-2 infection is a multistep process that involves binding to ACE2 via the surface S protein, employment of the cellular serine protease TMPRSS2 for S protein priming, and fusion between the cellular and viral membranes for cell entry.38-40 Viral genomic RNA is consequently released and translated into viral polymerase proteins, viral RNA, and the proteins are replicated, transcribed, translated, and assembled to form mature virions that are finally released from the host cell.38-40 The severity of the clinical manifestation of SARS-CoV-2 infection depends on a variety of factors which, in addition to the viral virulence and load, include the efficiency of the innate and adaptive immune response.2,41,44 The results of the reported studies suggest that OM-85 is effective in inhibiting SARS-CoV-2 infection in several experimental models by interfering with various step of the infection cycle. Pivniouk et al.13 showed that, downregulation of ACE2 and TMPRSS2 transcription on epithelial cells by OM-85 decreased virus attachment to the host cell membrane and SARS-CoV-2 S1 protein–mediated cell entry. Moreover, they demonstrated that the OM-85 induced reduction of ACE2 and TMPRSS2 transcription in lung cells was Myd88/Trif dependent. Interestingly, it was previously shown that the involvement of the Toll-like receptor adaptors MyD88 and Trif are required to promote the production of interferon-β by OM-85 activated bone marrow-derived dendritic cells.16
Type I IFN T are critical cytokines for protection against viruses during the acute phases of the infection, directly suppressing viral replication, thereby activating the immune responses.43 Interestingly, in the experiment evaluating epithelial cell infection, they further demonstrated the key event characterising the OM-85–induced suppression of SARS-CoV-2 infection is the reduction of ACE2 expression. The OM-85-induced suppression of epithelial cells infection obtained using SARS-CoV-2 pseudotyped particles were confirmed by experiments performed with live SARS-CoV-2 obtained from the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA). The ‘anti-SARS-CoV-2’ activities of OM-85 were evaluated on human bronchial epithelial cells by Fang et al.,14 who reported the inhibition of the transcription and of the expression of ACE2, TMPRSS2, but also of DPP4 proteins. The DPP4 serine exopeptidase, also known as CD26, is involved in multiple physiological processes, including the cleavage a broad range of substrates, such as growth factors, chemokines, neuropeptides, and vasoactive peptides.18 Recent evidence suggests that human DPP4/CD26 may interact with the S1 domain of the SARS-CoV-2 S glycoprotein and that DPP4 modulation or inhibition may prevent infection and/or progression of COVID-19.44 There is also evidence suggesting that DPP4 inhibitors could prevent an excessive inflammatory response, thereby reducing the production of pro-inflammatory cytokines, and exert anti-fibrotic activity.41 These properties may be of potential use for halting progression to a hyper-inflammatory state associated with severe COVID-19.
Fang et al.14 also showed that OM-85 treatment was effective in upregulating ADAM17, HPSE and HA production in human bronchial epithelial cultures. Upregulating ADAM17 expression, OM-85 promoted the cleavage of the ACE2 from the epithelial cell membrane with release in the extracellular milieu of a high amount of that protein in the soluble form. sACE2 may compete with membrane-anchored ACE2 receptors for binding sites of the SARS-CoV-2 virus S proteins, and act as a soluble decoy that can broadly block extracellular virus particles with limited potential for viral escape.45 Also, the soluble form of heparan sulfate, whose release is promoted by OM-85, can act as a target decoy, and contribute to the inhibition of epithelial cell infection.46,47
Finally, OM-85 also induced synthesis of hyaluronic acid which can inhibit virus cycle replication at different stages, including internalisation and transcription phases, and even by directly killing the virus.48 The ability of OM-85 to counteract CoV infection through induction of an effective immune response was evaluated in a mouse model by Salzmann et al.15 They demonstrated that oral OM-85 treatment reduced a MCoV lung infection via the promotion of lung macrophage anti-viral functions through an early IFN signalling activation. In OM-85 treated mice, massive reduction of viral load was associated with reduced apoptotic burden in lung tissue, and with an early recruitment of lung macrophages. The recruitment was associated with a phenotypical change characterised by increased mRNA levels of genes related to antigen presentation already detectable before exposure to MCoV infection and a faster activation of the IFN system genes in the first 2 days following infection in OM-85-treated animals. This rapid ‘innate immunity training’, leading to lung macrophage pre-activation, was able to enhance viral clearance and to reduce the virus-induced lung damage, confirming the immunomodulatory properties of OM-85. Finally, transplantation of naïve macrophages, and monocytes to recipient mice which were consequently infected with MCoV resulted in enhanced IFN signalling and, thereby, a faster viral resolution, suggesting that the overall number is even more essential than an additional pre-activation for a fast recovery. Due to the similarities between the MCoV used in this model and human COVID-19, it is possible to speculate that similar mechanisms, i.e., oral training of the immune system using OM-85, could be beneficial also in SARS-CoV-2 infection.
CONCLUSION
In experimental model of respiratory infections, OM-85 has the capacity to modulate both the innate and the adaptive immune response, conferring efficacious broad Ig-related and cell-mediated immunity to the respiratory system. The outcomes of the experimental studies reported in this review support the hypothesis that OM-85 may aid in preventing SARS-CoV-2 infection and/or reduce COVID-19 severity by inhibiting multiple steps of SARS-CoV-2 cell infection and by its immunomodulatory properties. The proven safety profile of OM-85 demonstrated by decades of clinical use as an immunomodulator,49 warrants future testing of this standardised bacterial extract in clinical trials to assess its potential as a complementary prophylactic and therapeutic approach in the current COVID-19 pandemic.