Abstract
Objective: The median length of survival for cystic fibrosis (CF) has significantly increased in recent years; however, this means that endocrine complications are more frequently observed in affected patients. This research was aimed at evaluating the frequency, clinical and paraclinical features, and the treatment of linear growth and pubertal disturbances, vitamin D insufficiency/deficiency, CF-related bone disease, and adrenal and thyroid function disturbances in patients with CF, as well as to explore their association with lung function and colonization.
Method: A cross-sectional study with a retrospective collection of information from patients under 18 years of age with a confirmed diagnosis of CF, evaluated on an outpatient or in-hospital basis by a pediatric pulmonologist or endocrinologist, or in a multidisciplinary CF consultation, between January 2011–December 2020.
Results: A total of 87 patients were included. A high frequency of endocrinopathies was found. Vitamin D insufficiency/deficiency was persistent despite cholecalciferol supplementation in 72%, short stature in 38%, pubertal delay in 26%, and thyroid axis disorders in 8% of cases. Bone disease involving densitometry and adrenal insufficiency secondary to chronic corticosteroid use were also frequent among the screened subjects (44% [n=16] and 12% [n=51], respectively). No significant differences were found for pulmonary function or bacterial lung colonization in the context of any endocrinopathies.
Conclusion: Endocrine comorbidities are frequent in patients with CF, and early recognition improves the prognosis and quality of life in these patients. The findings for this cohort ratify the importance of multidisciplinary management of patients with CF.
Key Points
1. The present study reveals that pediatric patients with cystic fibrosis (CF) have a high prevalence of endocrine abnormalities that should be recognized early by treating clinicians.2. This article describes the prevalence, clinical and paraclinical features, and management of linear growth and pubertal disorders, vitamin D insufficiency/deficiency, CF-related bone disease, and adrenal and thyroid dysfunction in patients with CF, as well as their association with lung function and colonization.
3. Endocrine abnormalities in pediatric patients with CF should sensitize clinicians to the need for early and timely multidisciplinary management to improve the prognosis and quality of life of affected patients.
INTRODUCTION
Advances in the medical and nutritional care of patients with cystic fibrosis (CF) have increased life expectancy in recent years; for this reason, more patients develop disease-related endocrine comorbidities.1 Globally, the presence of endocrine disruption is associated with a negative impact on the clinical outcomes and mortality of those affected.2
There is still not enough data to show the frequency of endocrinopathies in the pediatric population. Therefore, the present study was aimed at evaluating the frequency of vitamin D insufficiency/deficiency, bone disease involving densitometry, thyroid and adrenal axis disorders, short stature, and pubertal delay in a population of children with CF in Medellín, Colombia. Additionally, the authors intended to describe the screening and management of these pathologies over the last 10 years, and explore the relationship between pulmonary function impairment, bacterial pulmonary colonization, and the presence of endocrinopathies.
METHODS
A cross-sectional study was conducted at the Hospital San Vicente Fundación in Medellín, Colombia. Patients under 18 years of age who were evaluated in a multidisciplinary CF consultation involving endocrinology, pulmonology, gastroenterology, and nutrition, or patients who had been assessed by pulmonology or pediatric endocrinology on an outpatient or in-hospital basis from January 2011–December 2020, were identified.
Only those who met the criteria for a confirmed diagnosis of CF were included in the analysis, which was defined as evidence of two CF-causing CFTR gene mutations in the molecular test, and/or positive results of two iontophoresis with direct Gibson and Cooke’s test >60 mmol/L, or indirect tests with values >80 mmol/L.3
An instrument was designed to collect the information, including sociodemographic aspects, assessment of pulmonary condition, frequency and characteristics of linear growth, pubertal disturbances, vitamin D insufficiency/deficiency, CF-related bone disease, and adrenal and thyroid function disturbances.
The socioeconomic stratum was evaluated according to the stratification system of the Colombian National Planning Department; classifying 1 and 2 as low, 3 and 4 as medium, and 5 and 6 as high.
Standard deviation (SD) values were used for anthropometric assessment according to World Health Organization (WHO) charts.4 Low height risk was defined as a height/age ≥-2 to <-1 SD, and low height <-2 SD. The nutritional status classification was assessed according to resolution 2,465 of 2016. For those <5 years of age, with weight/age z-score: moderate undernutrition <-2 to ≥-3 SD, and severe undernutrition <-3 SD; and for ≥5 years with BMI/age z-score: risk of thinness ≤-1 to ≥-2 SD, and thinness <-2 SD. Mammary and testicular development were assessed by the Tanner scale (pre-pubertal <2 and pubertal ≥2).5,6
Lung function impairment was evaluated in children ≥6 years by forced expiratory volume in the first second (FEV1) measured by spirometry. Percent predicted values of FEV1 of 70–80% were considered mild, 60–70% moderate, 50–60% moderate-severe, <50% severe, and 35–50% very severe.7
Pulmonary colonization was defined as the presence of two or more cultures of respiratory secretions positive for one or more micro-organisms.8
Bone mineral density (BMD), measured by bone densitometry (DXA) of the total body without head and lumbar spine adjusted for age, gender, and height, was evaluated in patients >8 years in accordance with the recommendation of the European Society for Clinical Nutrition and Metabolism (ESPEN), the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN), and the European Cystic Fibrosis Society (ECFS) guidelines.9
The following definitions were established for the classification of endocrinopathies:
Delayed puberty: absence of thelarche in young females at 13 years of age, and absence of testicular volume of 4 ccs or greater in young males at 14 years of age.10
Vitamin D deficiency: serum 25-hydroxyvitamin D (25[OH]D) concentrations <20 ng/mL.11 Vitamin D insufficiency: serum 25(OH)D concentrations between 20.0–29.9 ng/mL.11
Bone disease: DXA of the lumbar spine or total body without head <-2DE of BMD z-score.9
Impaired thyroid function: Thyroid-stimulating hormone values persistently above the upper limit of the reference range for age associated with low T4L (primary hypothyroidism) or normal T4L (subclinical hypothyroidism).12
Adrenal insufficiency: 8 am cortisol level <3 μg/dL. Those with levels between 3–10 μg/dL were classified as indeterminate, and a low-dose adrenocorticotropin (ACTH) stimulation test was requested. A cortisol peak <18 μg/dL was considered diagnostic of adrenal insufficiency.13
Risk of adrenal insufficiency due to prolonged glucocorticoid use: oral corticosteroid use at supraphysiologic doses (>12 mg/m2/day of hydrocortisone equivalent) for >2 consecutive weeks or >3 weeks cumulative over the past 6 months, or inhaled corticosteroid (ICS) use at high doses (beclomethasone dipropionate >400 mcg/day, budesonide >400 mcg/day for children up to 11 years of age, and >800 mcg/day for those >11 years, fluticasone propionate >500 mcg/day) for more than 1 year.13,14
The data obtained were recorded in Microsoft Excel (Windows7®️, Redmond, Washington, USA) and subsequently analyzed with R Project version 4.1.3. To explore the association between lung colonization and lung function with the different endocrinopathies, χ2 association tests or Fisher’s exact test were performed. A P-value of less than 0.05 was considered significant.
The protocol of the study was approved by the local ethical board.
RESULTS
A total of 386 histories were reviewed, of which 87 met the inclusion criteria. A higher percentage of affected males were found with CF, usually with pre-pubertal Tanner stage, along with a mean z-score for risk of short stature (Table 1). Regarding nutritional status, 6.2% of children <5 years, and 21% of children ≥5 years had malnutrition.
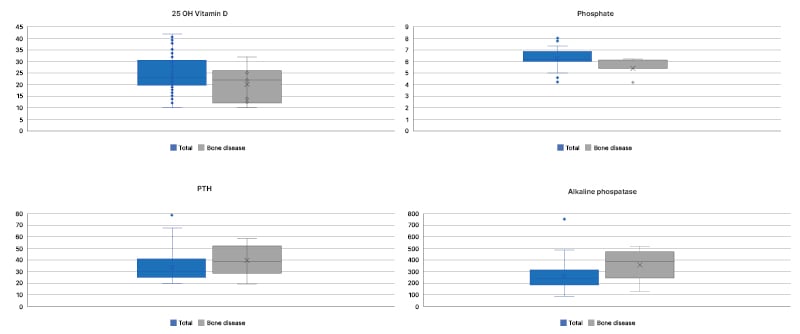
Figure 1. Frequency distribution of serum 25-hydroxyvitamin D, phosphate, parathyroid hormone, and alkaline phosphatase levels in the total population compared to children with bone disease measured by bone densitometry.
25 OH Vitamin D: serum 25-hydroxyvitamin D; PTH: parathyroid hormone.
Concerning pulmonary involvement, 10% of the patients had a permanent requirement for oxygen. Of the 65 patients ≥6 years, 58 exhibited spirometry with a mean FEV1 of 70% (±20.4).
Delayed puberty was assessed in young females ≥13 years and young males ≥14 years; it was observed more frequently in males than in females, with a 3:1 ratio. Short stature was also observed more frequently in males (20 males [60.6%] versus 13 females [39.4%]).
The authors measured 25(OH)D levels in all patients, 8 am cortisol levels in 51 patients, and a stimulation test with low doses of ACTH in three children who had 8 am cortisol levels <3. An indeterminate result was obtained in the 8 am cortisol test in 19 patients, of whom 15 were at risk of adrenal insufficiency due to prolonged use of glucocorticoids. An adrenocorticotropic hormone (ACTH) stimulation test was not performed in any of them for confirmation.
Of the 49 patients >8 years old, 16 underwent DXA, with greater BMD involvement of the total body without head (n=6) for the lumbar spine (n=4), and no pathological fractures were observed in any patient.
The phosphocalcic profile was evaluated in all patients with serum levels of 25(OH)D, calcium, phosphorus, and alkaline phosphatase. Intact parathyroid hormone levels were assessed in 78 patients. Mean vitamin D levels were 24.9 ng/mL (±7.73 SD) with insufficiency/deficiency in 63 patients (67% with insufficiency and 33% with deficiency), all of whom were receiving cholecalciferol supplementation, with a median dose of 2,000 international units per day (interquartile range: 1,000–3,000). All patients showed exocrine pancreatic insufficiency, defined as the presence of pancreatic enzyme replacement. The mean calcium was 9.6 mg/dL (±0.69 SD), phosphorus 5.29 mg/dL (±0.59 SD), alkaline phosphatase 256.6 U/L (±103.3 SD), and intact parathyroid hormone 33.57 pg/mL (±12.73 SD). These values were compared with patients who were presented with bone disease measured by DXA (Figure 1).
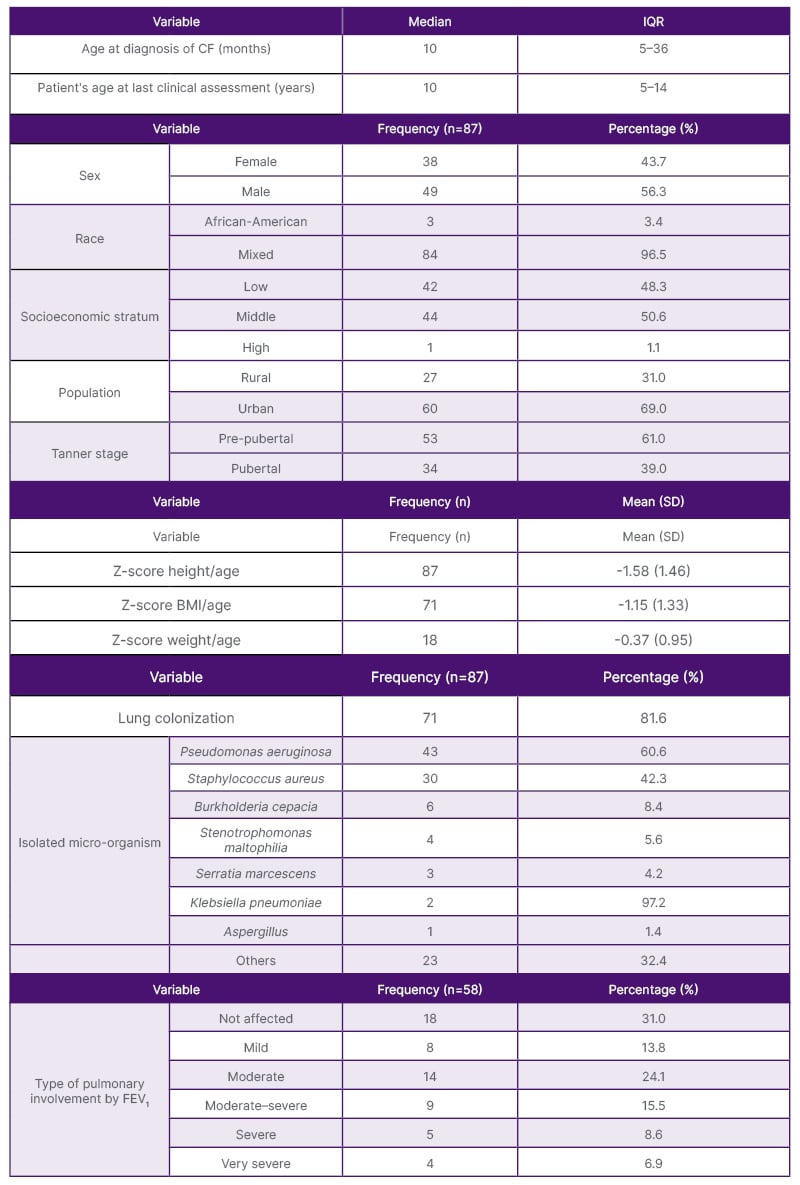
Table 1: Sociodemographic and clinical characteristics.
CF: cystic fibrosis ; FEV1: forced expiratory volume; IQR: interquartile range; SD: standard deviation.
The association between the endocrinopathies with bacterial lung colonization is presented in Table 2.
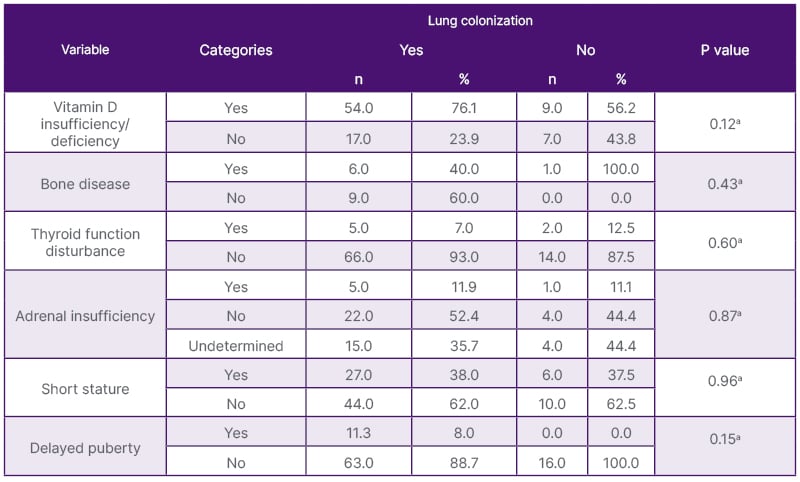
Table 2. Endocrinopathies according to lung colonization.
a: Fisher’s exact test.
Regarding the treatment of short stature, three patients had biochemical confirmation of hormone deficiency according to a clonidine test. They received recombinant human growth hormone at a median dose of 35 mcg/kg/day. Of the seven patients diagnosed with hypothyroidism, six had subclinical hypothyroidism, five received levothyroxine supplementation due to hypogrowth (short stature with poor growth velocity), and one patient had primary hypothyroidism.
Of the 87 patients, 70 (80.5%) received inhaled or systemic corticosteroid therapy, six received supraphysiological doses of oral corticosteroid for more than 2 consecutive weeks, 40 (46%) received a high-dose ICS for more than 1 year, 30 (75%) received beclomethasone dipropionate, seven fluticasone propionate, and three budesonide.
DISCUSSION
In the present study, the authors found a high frequency of endocrinopathies in the population of CF patients evaluated, with a frequency similar to that reported in studies worldwide.1,15 Although not completely comparable given the disparity in population and methodology, the findings agree with other authors who have reported that vitamin D insufficiency/deficiency was most frequently found in endocrinopathy. In a retrospective study evaluating 101 children, adolescents, and young adults with CF, vitamin D insufficiency and deficiency, defined as 25(OH)D concentrations <30 ng/mL and <11 ng/mL, were documented in 90% and 7% of patients, respectively.16 In this cohort, deficient vitamin D levels were more frequently described, possibly because a higher cut-off point was used to define vitamin D deficiency (<20 ng/mL). Similar results were reported by Aziz et al.17 who retrospectively evaluated 69 children with CF: 40.5% were vitamin D deficient and 31.8% were insufficient, for 72% of the population with vitamin D levels below 30 ng/mL. However, there was a discrepancy among the results of different studies secondary to multiple factors determining suboptimal vitamin D levels, such as deficient nutritional intake, decreased outdoor activity, altered vitamin D hydroxylation, corticosteroid use, and decreased intestinal absorption secondary to exocrine pancreatic insufficiency.18 In this population, all subjects had exocrine pancreatic insufficiency, with a significant percentage of patients reported with malnutrition, and chronic glucocorticoid use.
The second most frequent endocrinopathy was short stature. In a study that evaluated growth status in 13,116 children (age range: 0–18 years) seen at Cystic Fibrosis Foundation-accredited centers in the USA in 1993, 22% were below the 5th percentile for height-for-age based on the National Center for Health Statistics (NCHS)/Centers for Disease Control (CDC) growth.19 Also, they observed significant sex differences in the incidence of stunting (height-for-age <5th percentile) during adolescence; young males aged 11–14 years showed a lower incidence of stunting (19%) than young females (29%), while the opposite trend was observed between the ages 15–18 (34% in male patients versus 28% in female patients).19 The authors found a higher frequency of short stature in this cohort despite presenting a lower percentage of malnutrition (21% >5 years of age and 6.2% <5 years of age) compared to the previous study, which reported weight-for-age <5th percentile in 47% of infants and 34% of adolescents.19 There could be a contribution of other growth-related factors that explain a higher frequency of short stature in this population, such as fat and micronutrient malabsorption, chronic infection, and chronic glucocorticoid treatment, which were frequent.19 In addition, some data support the relationship between short stature and pubertal delay in the male population. Landon et al.20 evaluated the growth rate and pubertal status of 54 adolescent and young adult males with cystic fibrosis, of whom 39% were below the 5th percentile for height, and 28% between the ages of 14–18 had delayed pubertal development.20 The frequency of short stature and delayed puberty in the authors’ study was similar to that described, with greater incidence in the male sex possibly explained by more significant involvement in malnutrition, which was three times higher in males than females. Newer data in more recent cohorts of patients with CF show normalization of pubertal timing that may be secondary to better nutrition.21 However, most of this study’s patients were from low and middle socioeconomic strata, with a significant proportion of rural residents with economic and administrative difficulties in accessing nutritional supplements, and insufficient caloric intake.
Although CF-related bone disease (CFBD) screening of the entire population was not performed, among children who were screened with DXA, CFBD was frequent. In a systematic review, less than 5% of children with CF had CFBD, but this proportion increased to 20% in adolescence, with 55–65% of patients >45 years of age affected.22 Several studies have reported decreased BMD in children and adolescents with CF.23-25 Furthermore, peak mineralization has been reported to be significantly reduced in adolescents.26 In the authors’ cohort, all patients screened with DXA were adolescents, had vitamin D insufficiency/deficiency, and were receiving chronic steroid therapy, which may have contributed to a higher frequency of bone disease in this population.
Concerning alterations in adrenal function, a retrospective study diagnosed iatrogenic adrenal insufficiency in 20.5% of their cohort. The use of high-dose ICS (beclomethasone >400 μg and fluticasone >500 μg per day), especially fluticasone, appeared to be the significant risk factor.27 The percentage of patients who were diagnosed with adrenal insufficiency in the authors’ cohort was lower (12%); however, this was similar to that reported by Préville-Ratelle et al.,28 who indicated that only 8% of the adult population with CF had adrenal insufficiency. This suggests a possible underdiagnosis secondary to difficulties in the recognition, and lack of systematic detection, of this entity.28 In the authors’ study, deficiencies in the screening of adrenal insufficiency were also evidenced. The main limitation was the difficulty in interpreting indeterminate results due to the lack of an ACTH stimulation test, which is the confirmatory test. In the authors’ environment, the lack of availability of the ACTH analog, due to a shortage, added to limited authorizations by the National Institute of Drug and Food Surveillance (INVIMA), hindering the generalized performance of this test.
Finally, the prevalence of thyroid dysfunction in patients with CF remains controversial. Some studies have reported normal thyroid function, while others have reported subclinical hypothyroidism.29,30 Serum triiodothyronine (T3) values are often decreased due to impaired conversion of thyroxine (T4) to T3 in secondary to acute and chronic illnesses that frequently occur in patients with CF. The authors findings reflect the presence of subclinical hypothyroidism in children with CF, but with lower frequency than that reported in previous studies, probably because of a lower prevalence of iodine deficiency in the authors’ study population. According to the latest report of the National Survey of Nutritional Status (ENSIN) 2015, iodine deficiency in Colombian children aged 1–4 years was 8.1%, and in children aged 5–12 years was 4.4%.31
One of the most critical limitations was the need for more information from unscreened patients, which contributed to the lack of a sample with non-significant findings. Nevertheless, it is important to consider that part of the lack of significant differences in the presence of endocrinopathies according to lung colonization may be due to small sample size, and lack of power to prove differences. In addition, the economic and administrative barriers that limited the performance of the requested studies, and the continuity of the established treatments in many patients, could have generated confusion bias by increasing or decreasing the prevalence of CF-related endocrinopathies. However, though limitations were found, this is the first study in the authors’ setting to show a high frequency of endocrinopathies in a population of children with CF.
CONCLUSION
Endocrine comorbidities are frequent in pediatric patients with CF. This investigation reinforces the importance of a multidisciplinary follow-up of pediatric patients with CF to identify affected children and those most vulnerable, and to take appropriate preventive treatment and follow-up measures to impact clinical outcomes, pulmonary function, and quality of life in this population group.