INTRODUCTION
In cases of pulmonary fibrosis attributable to interstitial lung disease, whether the aetiology is known or unknown, the presence of at least two out of three criteria defined as worsening respiratory symptoms, functional impairment, and radiological progression within the past year characterises progressive pulmonary fibrosis (PPF).1-3 There are several diseases that can lead to PPF, and there is insufficient experience with antifibrotic treatment in patients other than those with idiopathic pulmonary fibrosis (IPF). This study aims to investigate the effectiveness of antifibrotic therapy in the treatment of cases with PPF other IPF.
METHOD
Patients followed up at the Interstitial Lung Diseases Outpatient Clinic of the Department of Chest Diseases, Istanbul University Faculty of Medicine, Türkiye, diagnosed with PPF according to the current guidelines, were included in the study.1,2 Patients under 18 years of age, those with transaminases or bilirubin levels >1.5 times the upper limit of normal, those with creatinine clearance <30 mL/min, and those with chronic liver disease were excluded from the study. Demographic characteristics, clinical features, comorbidities, blood oxygen saturation (SpO2), 6-minute walking distance (6MWD), spirometry, and diffusing capacity measurements of patients using antifibrotics for at least 6 months were recorded. Functional parameters at 6 months and 1 year of treatment were compared with baseline data.
RESULTS
In accordance with the guidelines, 105 cases diagnosed with PPF and under antifibrotic treatment were included in the study. Thirty-seven percent of the cases (n=39) had systemic sclerosis, 16% (n=15) had hypersensitivity pneumonitis, 11% (n=12) had rheumatoid arthritis, and in 37% (n=38) of the cases there was lung involvement associated with other systemic diseases (Table 1). The majority of cases (n=91; 86.7%) were using nintedanib, while 14 cases were using pirfenidone. The most common side effect associated with nintedanib use was diarrhoea (n=10; 10%), followed by elevated liver function tests (n=12; 11%). Treatment was discontinued in eight cases with treatment-resistant weight loss, and they were switched to pirfenidone therapy. Four cases discontinued medication therapy during follow-up at their own request. Fifteen cases (14.3%) were lost during the course of antifibrotic use. At 6 months, functional evaluation was available for 58 cases, and at 1 year, it was available for 36 cases. In the pre-antifibrotic treatment evaluation, the mean forced vital capacity (FVC) was 1975.05±650.598 mL (range: 730–3,800), FVC% was 65.367%, diffusing capacity for carbon monoxide (DLCO) was 47.15±14.796% (range: 17–80), and 6MWD was 403.89±89.148 m (range: 105–600). At the 6-month mark, the mean FVC was 2079.12±682.368 mL (range: 730–3,500), FVC% was 67.22% (range: 23–143), DLCO was 49±14.2% (range: 17–85), and 6MWD was 443.43±72.629 m (range: 260–550) (Table 2).
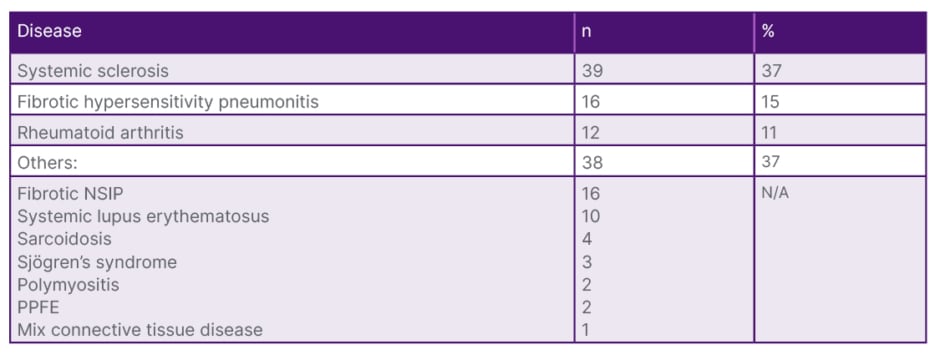
Table 1: Subgroup diseases of the patient population.
N/A: not applicable; NSIP: non-specific interstitial pneumonia; PPFE: pleuroparenchymal fibroelastosis.

Table 2: Functional changes of the study population after treatment.
6MWD: 6-minute walking distance; DLCO: diffusing capacity of the lungs for carbon monoxide; FVC: forced vital capacity.
At the end of 1 year of treatment, FVC was determined to be 2051.53±619.833 mL, FVC% was 68.22±22.271% (range: 33–145), DLCO was 48.53±16.168% (range: 12–80), and 6MWD was 424.94±97.680 m (range: 47–583). When compared with baseline values, there was no significant loss in FVC and DLCO at 6 months and 1 year. However, there was a significant increase in 6MWD (p<0.001 at 6 months; p<0.001 at 1 year). Except for the 12 cases identified with exertional desaturation during the pre-treatment functional assessment, no new cases with exertional desaturation were observed during follow-up.
CONCLUSION
No significant functional changes were observed in the 6-month functional assessment of PPF cases under nintedanib treatment. Our results suggest that rapid functional decline can be prevented with nintedanib therapy. In light of the available data, large-scale prospective studies are needed to demonstrate the efficacy of antifibrotic treatment in different PPF subgroups.4