Abstract
Background: Ovotesticular disorders of sex development (ODSD), previously known as true hermaphroditism, is a rare disorder of sexual differentiation that causes ambiguity in external genitalia and the presence of both ovarian and testicular elements in the gonads. The condition is characterised by the existence of testicular tissue with distinctive tubules and ovarian tissue in the same gonad (ovotestis), or separately in an individual. The most common cytogenetic karyotype is 46,XX. The age and presentation of symptoms vary, but typically include hypospadias, urogenital sinus, inguinal hernia, cryptorchidism, and gynaecomastia during the peripubertal age, as well as lower abdominal mass.
Case Presentation: A 15-year-old female exhibited typical male genitalia, including a normal penile shaft, scrotal sac, palpable testes, and a urethral opening, with a separate vaginal opening located between the penile shaft and scrotum. Pelvic MRI confirmed the presence of both testes within the scrotal sac and an infantile-sized uterus with a central endometrium, as well as a left-sided ovary in the pelvic region. The right ovary was not seen. Correlative ultrasound confirmed these findings and indicated that the testes had a normal echo texture and vascularity. The patient was diagnosed with ODSD, and further hormonal testing was recommended.
Conclusions: ODSD is a rare cause of genital ambiguity. Typically, both paramesonephric and mesonephric duct derivatives are seen, and most of the affected individuals present as neonates or infants with ambiguous external genitalia. At peripubertal age, the symptoms typically include hypospadias, urogenital sinus, inguinal hernia, unilateral or bilateral cryptorchidism, and gynaecomastia. Fertility is not uncommon and has been reported in female-reared patients. The ovotestis is the commonest gonad in people with ODSD and are known to be predisposed to germ cell tumours. Over 500 cases of ODSD have been reported in the literature. Ultrasound is the first line of investigation used to see the pelvic organs. The non-invasive test of choice for detecting paramesonephric and mesonephric duct derivatives is an MRI pelvis. The purpose of this paper is to describe various clinical and radiological features associated with ODSD.
Key Points
1. A rare congenital disorder called ovatesticular disorders of sex development (ODSD) is characterised by the co-existence of ovarian and testicular tissue in the same person. It is frequently accompanied with ambiguous external genitalia, and can manifest in a variety of ways with a range of symptoms, including gynecomastia, hypospadias, urogenital sinus, inguinal hernia, and cryptorchidism.
2. A combination of clinical, radiographic, and hormonal tests is frequently used to diagnose ODSD. To see the reproductive organs within and find paramesonephric and mesonephric duct derivatives, ultrasound and MRI are useful imaging tools. Hormone testing aids in evaluating hormone levels and choosing the best course of action.
3. Endocrinologists, geneticists, paediatric surgeons, and psychologists are required to manage ODSD, and treatment options include hormonal therapy to induce secondary sexual characteristics consistent with the patient’s gender identity, and surgical interventions to address ambiguous external genitalia. Long-term follow-up care, including hormone levels, bone health, and psychosocial support, is essential for people with ODSD.
INTRODUCTION
Ovotesticular disorders of sex development (ODSD) is an extremely rare congenital disease characterised by the presence of both testicular and ovarian tissue in the same gonad or person.1 In the literature on ODSD, approximately 500 cases have been reported.2 Hungerford et al.3 reported the first chromosomal findings in ODSD in 1959, when they discovered a 46,XX chromosomal complement in an individual’s peripheral blood lymphocytes with the disorder. ODSD is a genetically heterogeneous condition, with 46,XX being the most common karyotype. Depending on the amount of testicular tissue present, the phenotype of the external genitalia can range from normal male to normal female.1 The majority of ODSD cases have a normal female karyotype.4
CASE PRESENTATION
A child born from a consanguineous marriage who was reared as a female and is now 15 years old demonstrated a slow emergence of symptoms, such as a deepening of voice, increasing facial hair, and primary amenorrhoea. Tanner Stage III pubic hair development and Tanner Stage II breast development were seen in the patient. A penile shaft with a regular urethral entrance, a scrotal sac with normally positioned palpable testes, and a separate vaginal opening between the penile shaft and scrotum were all found on physical examination. Bilateral testes were totally descended and non-retractile. Following an investigation, it was discovered that the patient had a non-institutional unsupervised delivery at home, and so no relevant paperwork detailing newborn assessment was accessible. No other family member had a comparable condition. Prader staging was not included in the referring physician’s clinical evaluation.
Pelvis MRI
MRI pelvis scans showed both testes within the scrotal sac with the right testis measuring 28x15x17 mm along with mild hydrocele. The left testis measured 28x15x19 mm. There was a small left-sided epididymal cyst measuring 6.7 mm in diameter (Figure 1A and B). The penile shaft and glans were visualised with normal-appearing corpora cavernosa and corpora spongiosum (Figure 2). Prostate or seminal vesicles are not appreciated at their anatomical location.
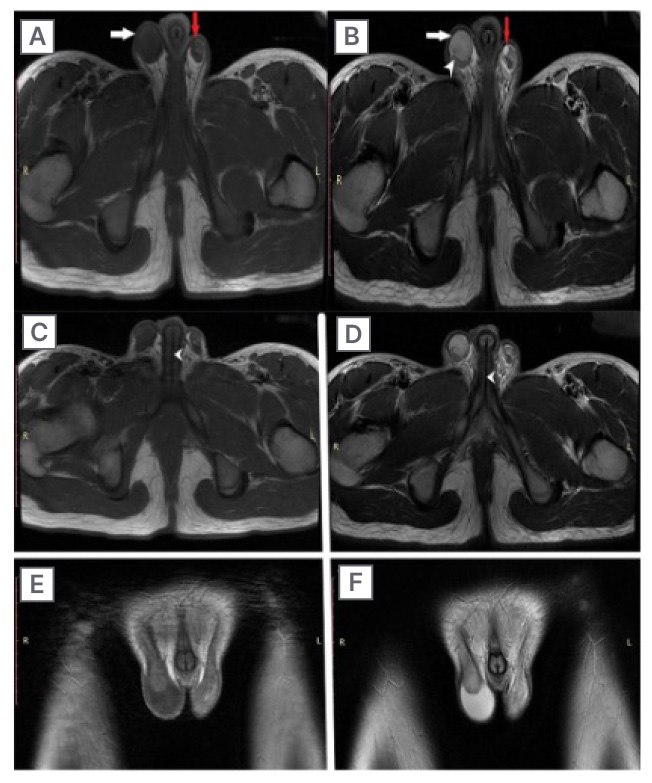
Figure 1: MRI showing the testicles of a 15-year-old individual with ovotesticular disorders of sex development, who presented with deepening of voice and primary amenorrhoea.
A and B) T2 and T1-weighted images showing testicles (white arrow), right-sided mild hydrocele (white arrowhead), and small epididymal cyst on left side (red arrow). C and D) Axial T1 and T2-weighted images showing the urethral opening (white arrow heads). E and F) Coronal T1 and T2-weighted images depicting scrotal sac with testicles.
Within the pelvis, there is an infantile-sized uterus measuring 24×12 mm in axial dimensions (Figure 2A and B). Central endometrium is appreciated measuring about 3.8 mm in thickness. The multi-cystic structure is seen in the left adnexal region conforming to the features of the ovary, measuring 25x19x14 mm (Figure 2). The right ovary was not seen. The uterine cavity continues inferiorly with the vagina, which has an external perineal opening. Within the perineum, the urethra opening into penile shaft is seen anteriorly with a separate vaginal opening posterior to it. Anal canal and anal verge were visualised separately (Figure 2D).
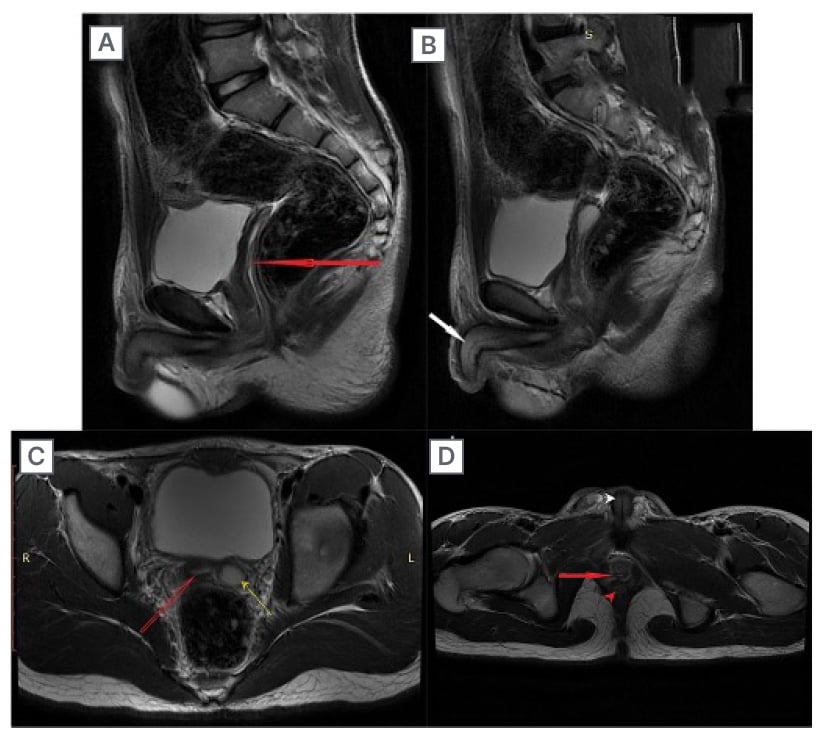
Figure 2: MRI showing the uterus with endometrium and penile shaft of a 15-year-old individual with ovotesticular disorders of sex development, who presented with deepening of voice and primary amenorrhoea.
A and B) Sagittal T2-weighted images showing the uterus with endometrium (red arrow in A) and penile shaft (white arrow in B). C) Axial T2-weighted image showing the uterus with endometrium (red arrow) and left sided ovary (yellow arrow). D) Axial T2-weighted image showing vagina (red arrow), anal canal (red arrowhead), and penile shaft with urethra (white arrowhead).
Greyscale and Doppler Ultrasound
A high frequency linear probe was used to perform an ultrasound of inguinoscrotal region. It revealed both testes with normal echotexture and normal blood flow on colour doppler (Figure 3).
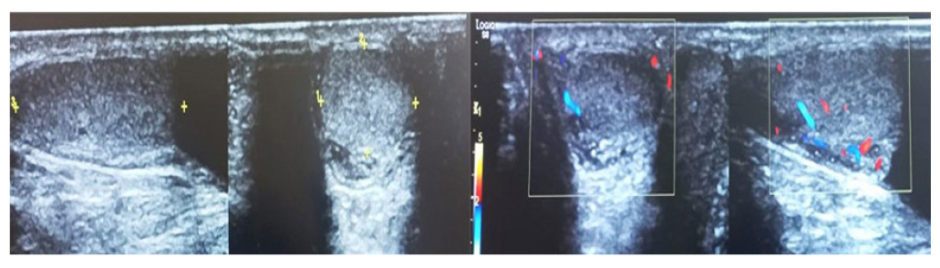
Figure 3: A greyscale doppler ultrasound of scrotum.
Biochemistry
Hormonal assessment was done resulting in the following parameters (Table 1).
No tissue was sampled for histopathological confirmation. Karyotyping was advised but could not be performed due to the lack of availability at the authors’ institution. Thus, on basis of the clinical, radiological, and hormonal investigations, a diagnosis of ODSD was made.
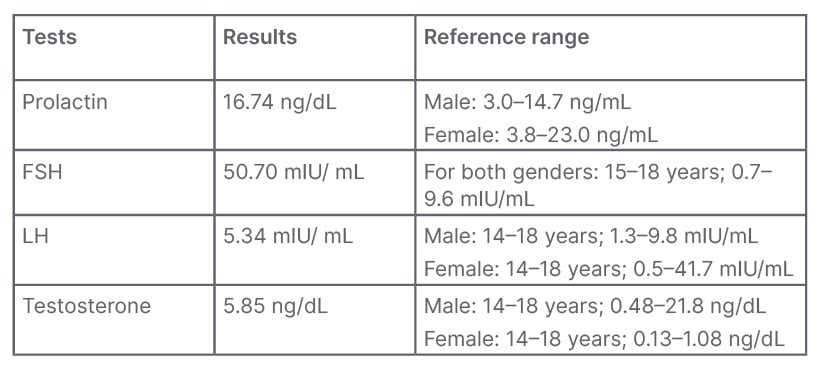
Table 1: Hormonal assessments.
FSH: follicle-stimulating hormone; LH: luteinising hormone.
DISCUSSION
Disorders of sexual development (DSD) is an umbrella term that refers to any problem in which the genital organ is atypical in relation to chromosomes or gonads. The most common causes of DSD are congenital adrenal hyperplasia and mixed gonadal dysgenesis, which account for more than half of all cases.5 The most uncommon form of DSD is ODSD, in which the external genitalia are ambiguous and the gonads contain both testicular and ovarian elements.6 The gonads can contain any combination of ovary, testes, or both (ovotestis).5 This anomaly can be caused by a variety of mechanisms, the most common of which is mosaicism or chimerism. Increasing maternal age is known to increase dizygotic twinning and, as a result, the risk of chimerism.6
The underlying karyotype frequently influences clinical presentation. In ODSD, the most common karyotype is 46,XX.7 The presentation features are variable. It also includes inguinal hernia or gynaecomastia, in addition to ambiguous genitalia. An ovary performs better than a testis in terms of functionality. The leydig cell function of the dysgenetic testis is generally insufficient, but ODSD may be well virilised, as in the authors’ case. Fertility is not uncommon and has been reported in female patients.2
In ODSD, the risk of gonadal neoplasm ranges from 2.6–4.6%, which is lower than in other DSD disorders.2 Ovotestis is the most common gonad in patients with ODSD, and these gonads are known to be predisposed to germ cell tumours.1 The most common histologic subtype of these tumours is dysgerminoma.2
In the literature, over 500 cases of ODSD have been reported. An MRI pelvis is the non-invasive test of choice for detecting paramesonephric and mesonephric duct derivatives. The pelvic organs can also be visualised using ultrasound.2 Additionally, radiology is important in the management of ODSD because it provides information on the presence and abnormalities of internal reproductive organs. Radiological studies, particularly ultrasound and MRI, assist clinicians in making an accurate diagnosis, determining the extent of the condition, and planning treatment options. As a result, radiology is an essential part of the diagnostic workup and treatment plan in patients with ODSD.
Differentiating ODSD from other illnesses with comparable clinical characteristics is critical for effective treatment. Androgen insensitivity syndrome, congenital adrenal hyperplasia, and mixed gonadal dysgenesis are the key differential diagnosis to examine. Androgen insensitivity syndrome is defined by individuals with a female phenotypic but a male karyotype (46,XY). These people are resistive to androgen activity, resulting in inadequate virilisation of the external genitalia.8 Congenital adrenal hyperplasia, on the other hand, is caused by enzyme defects in the adrenal steroid production. It can cause virilisation of the external genitalia in people who are born female. To distinguish congenital adrenal hyperplasia from ODSD, a complete clinical examination, hormone study, genetic testing, and radiological imaging are required.9 Mixed gonadal dysgenesis can manifest with a wide range of phenotypic characteristics, including ambiguous genitalia, and is distinguished by the presence of a streak gonad, a nonfunctional gonad, as well as a dysgenetic testis or ovary.9
To achieve the best results, ODSD requires a multidisciplinary approach, comprising of endocrinologists, geneticists, paediatric surgeons, and psychologists. Hormonal therapy and surgical treatments are two treatment possibilities. Hormonal therapy attempts to instil secondary sexual characteristics that are consistent with the patient’s gender identity. When a female patient is assigned, oestrogen replacement medication can be started. Testosterone replacement therapy may be considered for male patients. Gonadotropin-releasing hormone analogues may also be used to postpone puberty, and to give the patient time to make informed decisions about their gender identity.10 Surgical operations are adapted to the demands and preferences of the individual. Gonadectomy (removal of gonadal tissue) is commonly advised to lower the risk of cancer and to prevent hormonal abnormalities. After thorough examination of aspects such as fertility potential, anatomical traits, and patient preferences, the decision on which gonad to keep (ovary or testis) is taken. To correct the ambiguous external genitalia and provide a more coherent appearance, reconstructive procedures may be undertaken.11 After adequate counselling, the authors’ patient was referred to their physician for further management.
Individuals with ODSD require long-term follow-up care. To guarantee optimal health outcomes, hormone levels, bone health, and overall physical well-being, patients with ODSD must be monitored. Furthermore, psychosocial support is important in assisting patients and their families in coping with the difficulties associated with DSD. Individuals can benefit from support groups, counselling, and psychological interventions throughout their journey.12
CONCLUSION
Finally, the case report presented here demonstrates an unusual radiological presentation of ODSD with both external genitalia. The presence of testicular tissue with recognizable tubules as well as ovarian tissue in the same individual led to the diagnosis of ODSD. This case emphasises the significance of identifying paramesonephric and mesonephric duct derivatives using non-invasive diagnostic tools, such as pelvic MRI and ultrasound. Even though ODSD is a rare condition, it should be considered as a possible cause in neonates, infants, and peripubertal individuals of genital ambiguity. Understanding the clinical and radiological review of ODSD can help to ensure that affected individuals are managed in a timely and appropriate manner.
LIMITATIONS OF THE CASE REPORT
Genetic testing, such as karyotyping and molecular testing, is critical for detecting and distinguishing various DSD problems. The case report’s lack of specific genetic study data impedes a comprehensive understanding of the underlying genetic disorders that contribute to ODSD. Histopathological examination of gonadal tissue is also required to demonstrate the existence of ovarian and testicular elements. There may be a lack of precise histopathological descriptions or the inclusion of histological picture in the case report, which could provide additional insights into the specific histological abnormalities identified.
When reading the case report’s findings and conclusions, it is critical to keep these limitations in mind. More research is needed to increase our understanding of ODSD and its best therapeutic techniques, including bigger cohort studies with extensive genetic analysis and histopathological examinations.






