Abstract
Background: Caudal regression syndrome is a group of defects with incomplete development of the terminal vertebral column, with an incidence of 1 in 60,000 live births.
Case presentation: A 12-year-old female presented with dull, cyclical suprapubic abdominal pain occurring every month for the past 4 years. She was operated on for an anorectal malformation at birth. On examination, she had a blind-ending vagina with labial hypoplasia. She was diagnosed with caudal regression syndrome Type I, based upon the author’s findings of left renal agenesis along with low-lying blind ending uterus and sacral hypoplasia. She underwent a hysterectomy with right-sided salpingectomy along with the creation of a neo-vagina using a segment of the sigmoid colon. She was discharged after an uneventful postoperative course.
Conclusion: The condition is irreversible; however, repair and reconstruction of each systemic defect is warranted under a multidisciplinary team.
Key Points
1. Caudal regression syndrome is a rare condition affecting 1 in 60,000 births, presenting with complex congenital abnormalities requiring multidisciplinary management for improved patient outcomes.2. This case report describes a 12-year-old Pakistani girl with caudal regression syndrome Type I, detailing surgical reconstruction techniques and multidisciplinary approaches to manage multiple congenital anomalies effectively.
3. Early diagnosis and individualised, multidisciplinary treatment strategies, including surgical reconstruction, can significantly enhance the quality of life for patients with caudal regression syndrome, despite the condition’s irreversibility.
BACKGROUND
Caudal regression syndrome (CRS) is a group of defects with premature development of the terminal vertebral column, with an incidence of 1 in 60,000 live births.1,2 The term CRS was first coined by Duhamel in 1964.1 The condition may present with a wide range of abnormalities including partial or complete sacral/lumber agenesis, lower limb deformities, anorectal malformations, and urogenital anomalies.1,3 It is also related to vertebral, anal, cardiac, trachea-oesophageal, renal, and limb defects (VACTERL) syndrome.3
The condition occurs due to complex dysraphism with aberrations in gastrulation and failure of notochord development. This defect is attributed to the failure of neurulation along with the failure of the formation of somites being closely linked to the development of the heart and other organs.3 Similarly, the proximity of the caudal neurons, spinal, hindgut, and other elements involved in the closure of the neural tube results in defects of the urogenital, limb, and gastrointestinal systems.1
CASE PRESENTATION
A 12-year-old female of South Asian origin presented to the paediatric surgical outpatient department with complaints of dull, cyclical suprapubic abdominal pain occurring every month for the past 4 years. Her past medical, birth, developmental, and drug history were unremarkable. Her parents were in a consanguineous marriage (marriage between first-degree relatives) and they had three children. Out of these three, the patient was the eldest child. Both of her siblings were healthy and did not report similar complaints. Her mother reported a single-stage procedure at 2.5 months of age. However, medical records were unavailable, and the authors believe, from the appearance, that it was for a recto-perineal fistula.
On examination, her vital signs were stable, with a pulse rate of 85 bpm, blood pressure of 97/65 mmHg, respiratory rate of 17 breaths per minute, temperature of 98 °F, and blood oxygen saturation of 99% at room air. Her weight was 38 kg, and her review of systems was unremarkable. Her oral exam showed a submucosal cleft of the soft palate, and on genital examination she had a blind-ending vagina with labial hypoplasia.
An ultrasound of the abdomen showed left renal agenesis along with a low-lying blind-ending uterus. The MRI of the pelvis showed a hyperdense area consistent with haematosalpinx, confirming the presence of blood in the right fallopian tube and an absent vagina along with sacral hypoplasia as seen in Figure 1. Her echocardiogram did not reveal any cardiac anomaly.
She was diagnosed with CRS (Type I) and a gynaecological consult was sought to assess for reproductive potential. Intraoperatively, a rudimentary uterus, measuring approximately 2 cm in its greatest dimension, with a right-sided haematosalpinx was identified. The left fallopian tube was absent. The cervix and part of the vagina were also absent (Figure 2). The patient’s exceedingly small rudimentary uterus, absent vagina, and limited reproductive potential as per the intraoperative gynaecology review led to the decision for a hysterectomy with a right-sided salpingectomy. Furthermore, the decision was made keeping in view that the absence of the cervix results in sub-fertility, a higher chance of miscarriages, and predisposes to ascending infections. Both ovaries were preserved so that in future the child may have the option of egg retrieval and surrogacy. A segment of the sigmoid colon was used as a conduit for the creation of a new vagina in an iso-peristaltic fashion. For the reconstructive procedure, the authors utilised a combined lower abdominal and perineal approach. A single lower abdominal incision was made. Approximately 4 cm of the sigmoid colon was used to create the vaginal canal. The segment of the gut was stitched in a discontinuous manner with absorptive sutures to the rudimentary vaginal canal, establishing an anastomosis. The upper end of the segment was closed in a blind-ending fashion, while the lower end was anastomosed to the rudimentary vaginal canal to ensure continuity.
She had an uneventful recovery and was discharged on the fourth post-operative day with regular follow-ups. The follow-ups were planned for every 3 weeks in the initial period to monitor for any vaginal stenosis.
It is to be mentioned that during the entire process, careful consideration was taken to keep the confidentiality of the patient safe, and proper written consent was taken.
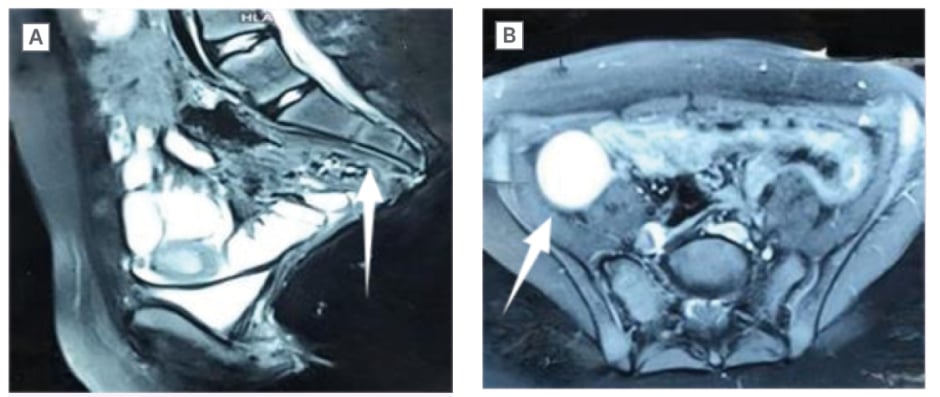
Figure 1: A) MRI showing sacral agenesis with hypoplastic L3, absent L4, L5, and coccygeal vertebrae. B) MRI showing right-sided haematosalpinx.
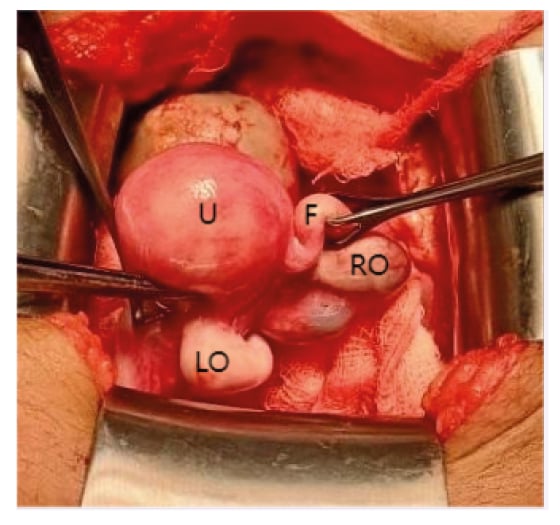
Figure 2: Operative image showing uterus, right fallopian tube, right ovary, and left ovary. There was absence of left fallopian tube, upper vagina, and cervix.
F: right fallopian tube; LO: left ovary; RO: right ovary; U: uterus.
DISCUSSION
CRS represents a spectrum of structural defects of the caudal region. The defects include premature termination of the vertebral column leading to partial or complete absence of sacrum and/or lumbar vertebrae.1 Along with that, affected persons may present with associated musculoskeletal anomalies such as dysplastic hips, underdeveloped musculature, club feet, gastrointestinal anomalies like anorectal malformations, oesophagal or duodenal atresia, certain neurological anomalies like defective bladder and bowel control leading to urinary and bowel incontinence, and varying degrees of motor and sensory loss. Urological anomalies may include renal agenesis or dysgenesis, hydronephrosis, renal ectopia, and genital anomalies like uterine abnormalities and hypospadias. Other associated anomalies include myelomeningocele, congenital heart defects, and facial anomalies such as cleft lip and cleft palate. Nonetheless, the children generally have normal neurodevelopmental outcomes.4
Although the aetiology is poorly understood and most cases are sporadic, a few factors have been identified that may contribute to the development of the defect. The three most important factors are maternal diabetes, vascular hypoperfusion, and genetic predisposition.5,6 Gestational hyperglycaemia is associated with alterations in the closure of the neural tube.7 The genotype of CRS is complex and multigenic with the caudal type homeobox 2 (CDX2) gene playing an important role in sacral morphogenesis.6 At the fetal level, it is believed to occur due to defects in the induction of the caudal elements in the embryo before the 28th day of gestation. This occurs secondary to injury in the posterior medial mesodermal axis causing the absence of development of caudal mesoblastic yolk.7
CRS is also associated with multiple congenital syndromes like VACTERL, OIES syndrome (Omphalocele, imperforate anus, cloacal exstrophy, spinal defects), and Currarino syndrome (a triad of sacral agenesis, presacral mass, and anorectal malformation).1,6
Several classification systems have been suggested for CRS; however, Renshaw provided a classic classification including five subtypes. Type I is total or partial unilateral sacral agenesis. Type II is variable lumbar but total sacral agenesis with the ilia that are articulating alongside the lowest vertebra. Type III is variable lumbar and total sacral agenesis with the caudal end plate of the lowest vertebra resting above either fused ilia or an iliac amphiarthrosis. Type IV is soft tissue fusion of both the lower limbs. Type V also known as sirenomelia or mermaid syndrome, where there is a single femur and tibia (fused bones of lower limb).8,9
The treatment of CRS involves multidisciplinary discussion and planning. The condition itself is permanent and repair of individual systems is the recommended approach. Each treatment option is tailored to the anatomical, pathological, and social aspects of the patient.1 Orthopaedic treatment includes spinopelvic stabilisation, correction of spinal deformity, correction of scoliosis/kyphosis, and contracture corrections.10 Neurosurgical treatment includes the release of conus in spinal cord tethering and surgery for myelomeningocele.11 Urological treatment includes treatment of neurogenic bladder and surgery for vesicoureteral reflux.12 Gastrointestinal treatment includes the correction of anorectal anomalies with the treatment of faecal incontinence.13 Depending on the type of malformations, patients are also offered plastic and reconstructive options for cosmetic reasons.14
Preserving fertility potential is a key aspect in the treatment of CRS. Based on the underlying anatomy, various options are available. The primary objective is to preserve reproductive and sexual function and to optimise fertility outcomes as much as possible.14
CONCLUSION
CRS is a rare and challenging condition, often presenting with a wide array of defects. The condition is irreversible; however, repair and reconstruction of each systemic defect is warranted under a multidisciplinary team. This case report highlights the unique aspects of the presentation and management of CRS, emphasising the importance of individualised treatment plans. By sharing this case, the authors aim to enhance the understanding of CRS and guide future clinical practice.






