Abstract
Pre-clinical models and human clinical trials have confirmed the ability of cancer vaccines to induce immune responses that are tumour-specific and, in some cases, associated with clinical response. However, cancer vaccines as a targeted immunotherapy strategy have not yet come of age. So, why the discordance after so much research has been invested in cancer vaccines? There are several reasons for this that include: limited tumour immunogenicity (limited targeted antigen expression, antigen tolerance); antigenic heterogeneity in tumours; heterogeneity of individual immune responses; multiple mechanisms associated with suppressed functional activity of immune effector cells, the underlying rationale for the use of immune checkpoint inhibitors; and immune system exhaustion. The success of checkpoint therapy has refocussed investigations into defining relationships between tumours and host immune systems, appreciating the mechanisms by which tumour cells escape immune surveillance and reinforcing recognition of the potential of vaccines in the treatment and prevention of cancer. Recent developments in cancer immunotherapies, together with associated technologies, for instance, the unparalleled achievements by immune checkpoint inhibitors and neo-antigen identification tools, may foster potential improvements in cancer vaccines for the treatment of malignancies.
INTRODUCTION
Cancer is a genetic and epigenetic disease of multicellularity, driving transformed cells to uncontrolled growth, invasion, and metastasis. In cancer, intracellular mechanisms controlling cellular proliferation are damaged first, which allows certain cells to progress to malignant transformation.1 Activation of oncogenes by various genetic alterations occurs later in the transformation process when genetic instability has reached a critical level.2 Understanding these molecular changes, along with their protein expression correlates, adds more precision to the anatomical classification of cancers and has ushered in the era of targeted therapy. This effort has led to the discovery of new targets and drugs, and the definition of new biomarkers.
The recent, dramatic success of immunotherapy for treatment of some of the most highly standard chemotherapy-resistant cancers (e.g., melanoma, lung cancer) has refocussed research on the role of the immune system as the main extracellular mechanism for cancer control. Using established databases, this new direction of research allows for the definition of different immune profiles across cancer types.3,4 Novel biomarkers, identified using simple immunohistochemistry (Immunoscore®) or multi-omic methods, have been defined and validated, as well as led to better prognostication that may surpass the traditional tumour, node, metastasis (TNM) staging system.3,5 A major focus of ongoing research is to determine why immunotherapies work or fail, and how they can be improved to reach their hoped-for potential as a broadly transformative treatment for cancer. Based on the presence of lymphocyte infiltrates and their types, three major immune phenotypes have emerged that correlate with response to immunotherapy: hot or inflamed, which respond well to immune checkpoint inhibitors (ICI); cold or ‘immune desert,’ which do not respond to ICI; and two subtypes within the altered (both excluded and immunosuppressed) immune phenotype. The immunosuppressed phenotype is also expected to respond to ICI as it has pre-existing activated immune cells in the tumour.6,7
It is these phenotypes that cancer-focussed vaccines aim to address. ‘Hot’ tumours often have a high mutational load and therefore are expected to express neo-antigens to provoke a strong immune response. Cold tumours, by contrast, are cancers that, for various reasons, have not been recognised or provoked a strong response by the immune system. Herein lies one of the limitations of immunotherapy. Characteristically hot tumours are limited and include bladder cancer, head and neck cancers, kidney cancer, liver cancer, melanoma, and non-small cell lung cancer, as well as tumours of different types with high microsatellite instability. The challenge is in the application of immunotherapy to cancers that are immunologically cold, such as glioblastomas, ovarian, prostate, and pancreatic cancer.8 One of the most important questions for the future of immunotherapy is to determine how to make cold tumours immunoresponsive.
STRATEGIES AND APPROACHES IN CANCER VACCINE DEVELOPMENT
Interest in targeted cancer immunotherapy by vaccination has been reinvigorated by the U.S. Food and Drug Administration (FDA) approval of ICI, which has had an impact on vaccine strategies for use in cancer therapy.9,10 Cancer vaccines, as a targeted immunotherapy strategy used for prevention (primary or secondary) or for treatment, have shown promise in preclinical animal studies, with the future aim of translation to the clinic. Some success is evident in the FDA approval of PROVENGE® (sipuleucel-T), a herpes simplex virus Type 1-derived oncolytic (T-VEC) immunotherapy that is injected directly into melanoma lesions; and in Gardasil®, which targets human papillomavirus known to cause cervical cancer. General research consensus is that a vaccine for cancer as a single entity is not practical because cancer reflects a myriad of different conditions.
The role of immunity in eradicating cancer is now considered in terms of stepwise events or the ‘immunity cycle’, a framework proposed by Chen and Mellman.6,11 This cycle identifies six steps preceding the killing of cancer cells by the immune system (Figure 1). Once the immune system is activated it is expected, based on clinical and preclinical studies, that immunosuppression would ensue to stop the immune attack against the tumour.7 Therefore, testing is underway of multiple strategies and approaches to activate the immune system for both hot and cold tumours (Table 1).
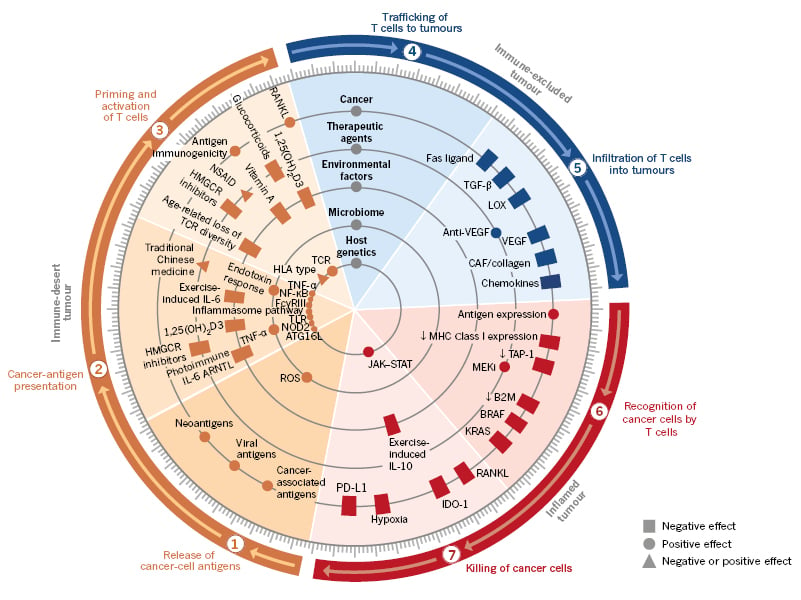
Figure 1: Multiple factors (tumour, host, and environment) affect each step of the immunity cycle.
The immune cycle can be correlated with the different immune phenotypes, and molecular and cellular abnormalities.
ARNTL: aryl hydrocarbon receptor nuclear translocator-like; ATG16L: autophagy related 16-like; B2M: β2 microglobulin; CAF: cancer-associated fibroblasts; FcγRIII: Fcγ receptor III; HLA: human leukocyte antigen; HMGCR: 3-hydroxy-3-methylglutaryl-CoA reductase; JAK–STAT: Janus kinase-signal transducer and activator of transcription; MEKi: mitogen-activated extracellular signal-regulated kinase inhibitor; MHC: major histocompatibility complex; NOD2: nucleotide binding oligomerisation domain-containing 2; NSAID: non-steroidal anti-inflammatory drugs; PD-L1: programmed death ligand-1; RANKL: receptor activator of nuclear factor κ-Β ligand; ROS: reactive oxygen species; TCR: T-cell receptor; TLR: toll-like receptors; VEGF: vascular endothelial growth factor.
Adapted from Chen and Mellman,11 used with permission.
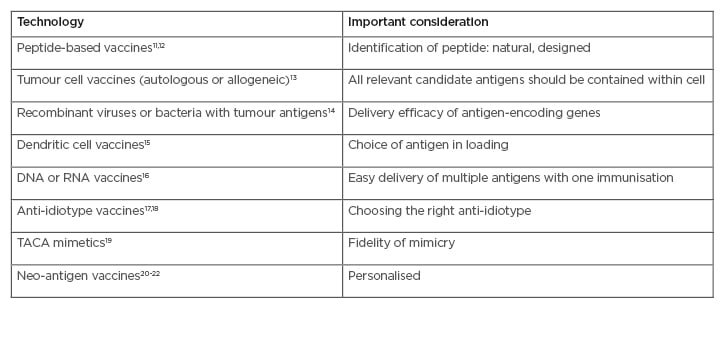
Table 1: Main types of cancer vaccines and adaptive immunotherapy.
TACA: tumour-associated carbohydrate mimetics.
Peptide-based vaccines rely on the development of molecular tools for improving, as well as studying, peptide-based vaccines.12 Whole-cell lysate vaccines are applicable to all patients, regardless of human leukocyte antigen (HLA) type.13 Recombinant DNA or viral vector-based vaccines focus on design, delivery, and combination strategies that break tolerance and generate a strong immune response.14 Dendritic cells are the most effective antigen-presenting cells for inducing T-cell proliferation, activation, and cross priming.15 Tumour-associated carbohydrate mimetics are peptides that mimic the carbohydrate three-dimensional configuration on certain cancer-related proteins.16,17
The anti-idiotype therapeutic vaccine racotumomab has been conditionally approved in Latin America as maintenance therapy for advanced non-small cell lung cancer.18 Recent breakthroughs in cancer immunotherapy demonstrate that clinical responses correlate with activation and expansion of tumour-specific T lymphocytes that mostly target mutation-based neo-antigens.19 Due to their economically effective, cold chain transport and lack of harmful ingredients, many antigens are in development as plant-based vaccines, with only a few undertaking clinical trials in humans.20,21
The discovery that cancer cells may evade the response of tumour-reactive T cells has ignited efforts to improve the efficacy of antitumour immune responses, with the hope of removing limits on the activation and maintenance of T-cell effector function.
Reprogramming a tumour microenvironment to trigger T-cell activation and enhance tumour immunity, in effect, making a tumour hotter, provides insight into enhancing immune response. A strategy in support of this involves targeting the checkpoint’s programmed cell-death protein-1 (PD-1) and programmed death ligand-1 (PD-L1) along with cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4). Developed ICI are negative-regulators of T-cell immune function but with different mechanisms of action.22
PD-1 is expressed on activated T and B cells, natural killer T cells, Type 2 innate lymphoid cells (ILC-2), and myeloid cells.23 CTLA-4 is expressed on Treg, activated T, and B cells. Expression of CTLA-4 on human natural killer T cells is unknown. As a result, CTLA-4 blockade disrupts the T-cell interaction with other antigen-presenting cells, such as dendritic cells, macrophages, or B cells, while anti-PD-1 blockade primarily blocks the tumour cell and cytotoxic CD8+ T-cell interaction.
Biomarker studies with anti-PD-1 and PD-L1, along with CTLA-4 clinical trials, support the hypothesis that these agents are most effective in patients who have pre-existing anticancer immunity. Determination of the basis of this pre-existing immunity may allow it to be utilised and amplified in vaccine strategies. ICI development demonstrated the concept of two nascent responses: first, the innate immune surveillance of cancer cells, and second, the adaptive immune response generated by the emerging tumour.
THE NASCENT IMMUNE SYSTEM
The immune system has evolved to distinguish self from non-self as a means to protect the host. A general feature of immune system mechanisms is that they detect structural features of non-self that mark them as distinct from host cells, reflecting a danger signal to the immune system.24 The emergence of cancer25 co-opts tissue-specific immune development to escape detection, augmented by failure of the immune system to perform its primary task of surveillance and elimination.26 However, there is evidence that innate and adaptive surveillance does occur. Natural anti-carbohydrate antibodies are known to mediate cancer cell death.27 Such natural antibodies, as part of innate immune surveillance, could promote tumour immunity by inducing immunogenic cell death, leading to immune priming and epitope spreading. Vaccine-induced anti-carbohydrate antibodies have displayed the same antitumour characteristics as natural anti-glycan antibodies.17 Thus, it may be possible to emulate innate antitumour responses in cancer therapies.
Burnet28-30 and Thomas31,32 hypothesised that the immune system can recognise nascent transformed cells, leading to elimination of the primary tumour formation.33 Tumours that are not eliminated undergo a process of immune editing,34 a reflection of the dynamic nature of immune surveillance, suggesting that at some point the antitumour immune surveillance was working, whether innate or adaptive. When the immune system is successful in eradicating incipient cancer cells, based on innate or adaptive immune response, no traces remain of its action. Mouse studies suggest that the immune system could initially see tumours as immunogenic, lending to a primed immune response.35-37 After this initial phase, and if the cancer is not eliminated, the immune system shuts down, which leads to cancer escape from immune surveillance. As a result, the failure to eradicate cancer is not a failure of immune priming. These observations have led to the hypothesis that immune sculpting may result in the emergence of a less-immunogenic clone that is undetected by the immune system or downstream suppressive mechanisms. These downstream suppressive mechanisms include immune checkpoints that may allow malignant cells to evade an effectively-primed immune response, hence the rationale for the emergence of ICI.22 It was 54 years following the research of Burnett and Thomas that anti-CTLA-4 therapies were approved.38
This leads to the question, where are we with enhancing the nascent response? Among clinical trials on clinicaltrials.gov, the search term “cancer vaccine” returned 58 early Phase I and combined Phase I/II, and 25 Phase II cancer vaccine trials that are actively recruiting. The distribution of these trials is listed in Table 2. The focus on T cells as the major immune effector mechanism for the action of cancer vaccines relies on processed tumour-directed peptides that activate T cells. This simplified view necessitates that, for acquired nascent immunity, the cancer-immunity cycle is initiated by the release of cancer cell antigens either shed by living cancer cells or released from dying tumour cells (see Figure 1). In either case, antigens are taken up and presented by antigen-presenting cells. Two antigen types are mainly represented in clinical trials in Table 2, with some considered as neo-antigens and others as tumour-associated antigens (TAA), incorporated into various platforms. Immune activity in cancer supports combining ICI with trials involving personalised tumour-specific neo-antigens and adaptive responses in general. However, currently there are 45 open vaccine trials without ICI, compared to 37 with ICI.
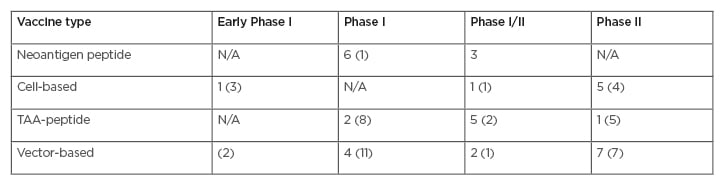
Table 2: Summary of open vaccine trials with and without checkpoint inhibitors.
Number in parentheses reflects the number of trials without immune checkpoint inhibitors combination.
TAA: tumour-associated antigen.
It is not clear whether cancer vaccines are part of a rational approach aimed at defined mechanisms. Systems vaccinology is an emerging field that applies omics technologies, in combination with bioinformatics tools such as transcriptional network analysis and predictive modelling, to study immune responses to vaccination.39 This integration to vaccine design requires the understanding of the molecular network mobilised by vaccination. What are the global correlates of successful vaccination, beyond the specific immune response to the antigens administered, for understanding the mechanisms that underlie successful immunogenicity? Functional genomics are being used to analyse specific molecular signatures and antigens, for use as predictors of vaccination efficiency. The immune response to vaccination involves the coordinated induction of master transcription factors that leads to the development of a broad, polyfunctional, and persistent immune response, integrating all effector cells of the immune systems.
TUMOUR MUTATIONAL BURDEN, NEO-ANTIGENS, AND THE PERSONALISATION OF IMMUNOTHERAPY
Significant past research has focussed on genetic abnormalities affecting cancer-related genes (oncogenes and tumour suppressor genes), to define oncogenic drivers, and select the most important ones for therapeutic targeting. The remainder of the mutations discovered through genomic analyses were considered irrelevant. With the advent of next generation sequencing, a multitude of mutations were detected, leading the field to consider tumour mutation burden and discover neo-antigens. Research highlighting the role of the immune system led to the discovery that efficacy of ICI was correlated with tumour mutation burden and the presence of these neo-antigens.40-44
Neo-antigens are specific to hot tumours and may be unique to each patient. As a result, the anticancer vaccine effort has sought to develop specific reactive T cells to these neo-antigens. Their diversity, however, makes adaptation for immediate clinical use more difficult, requiring complicated platforms capable of rapid sequencing of the patient’s genome to determine the most likely neo-antigens to be produced and given to the patient.45-47
The first step in the development of neo-antigen vaccines is the definition of the cancer mutanome for a specific patient.48 The availability of high performance platforms for next generation sequencing allows the rapid identification of tumour mutations in comparison to matched healthy tissue samples. Alterations that are likely to result in immunologically meaningful mutations are single nucleotide variations, gene fusions, frame shifts by small insertions or deletions, and cancer-associated epigenetic aberrations49 (at the transcriptional, translational, or post-translational levels). The second step is selection of the best neo-epitopes for vaccine design. Computational models have been developed to achieve this goal.50-53 The third step is to select the format of delivery of the vaccine. Commonly used formats include long peptides and RNA. Others, such as DNA plasmids, engineered bacteria or viruses, and antigen-loaded dendritic cells, are under consideration.48 The fourth step is to select the clinical setting for therapeutic application. Current practice suggests that these vaccines would work best in the adjuvant or minimal residual disease settings.
Some of these tumour-specific neo-antigens are known to be, or expected to be, common across a subset of patients and are called shared neo-antigens. Clinical trials of neo-antigens are shown in Table 3,54-66with some trials making use of typical peptide formulations while others involve a DNA or plasmid format. Neo-antigens such as tumour-specific antigens (TAA) are considered more immunogenic compared to self-antigens. TAA are now considered less favourable as vaccine candidates for several reasons: 1) being shared with normal tissues; 2) immune tolerance; and 3) heterogeneity within the same tissue, and among patients. However, heterogeneity may also occur with neo-antigens due to intratumour heterogeneity. Vaccines encoding xenoantigens, ‘non-self’ proteins that are highly homologous to their autologous counterparts, have been investigated as a means to increase immunogenicity and overcome tolerance to ‘self’ antigens.67 Likewise, mimotopes or vaccines that incorporate peptide mimics of tumour antigens can function by eliciting increased numbers of T cells that cross react with the native tumour antigen.68Mimotopes, which are xenoantigens, can function like neo-antigens in inducing immune responses because they are different from self-antigens. Mimotopes have broad applications.
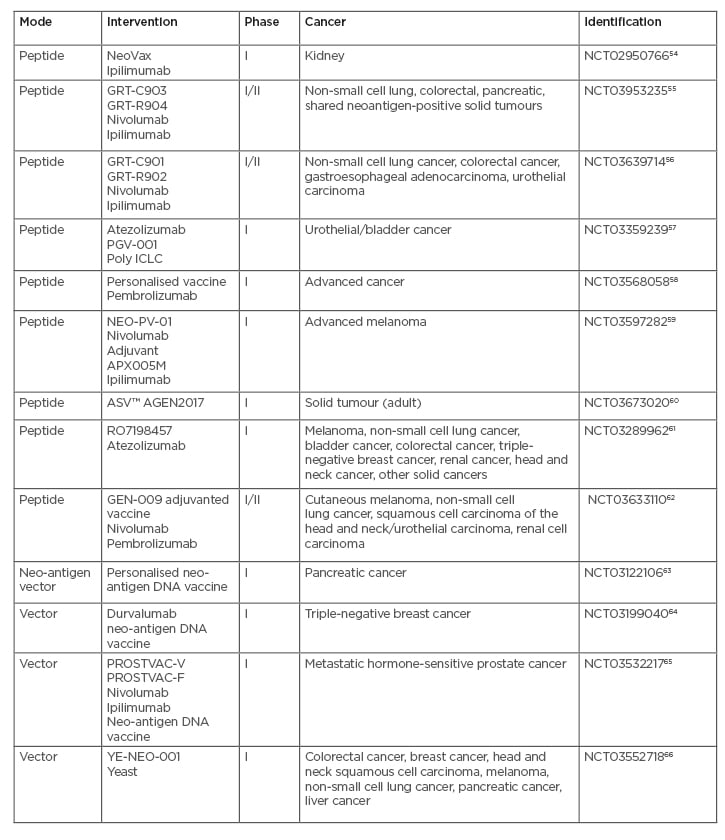
Table 3: Open clinical trials with neoantigen formulations.
Poly ICLC: polyinosinic-polycytidylic acid-poly-l-lysine carboxymethylcellulose; YE-NEO-001: neoepitope yeast vaccine.
For example, one mimotope of tumour- associated carbohydrate antigens is currently undergoing clinical testing.16,17 Mimotopes could be utilised to facilitate responses to cold tumours by recruiting TAA cross-reactive T cells and antibodies.
Presence of neo-antigens alone does not completely trigger an effective immune response; how the new antigens are presented also plays a role. For example, the presentation of a neo-antigen in low quantities, and with progressive minor modifications, may lead to immune tolerance rather than immune rejection.69
Conversely, high immunogenicity can still curtail a ‘one size fits all’ vaccine because of inherent heterogeneity in mutational rates. Neo-antigen vaccines are considered a means to enhance the nascent adaptive response, but to do so requires a vaccine to be developed from neo-antigens for every patient. This challenge may suggest an alternative strategy of using a whole-cell approach from a patient’s own tumour, to customise or tailor a personalised vaccine.
CANCER METABOLIC STRESS AND RESISTANCE TO IMMUNOTHERAPY
Immune escape in cancer may occur in early stages of the immune response, as the cancer undergoes immune editing and becomes invisible to the immune system. Activating mutations of certain oncogenes (KRAS, BRAF, or MAPK) may result in decreased expression of major histocompatibility complex class-1 (MHC-I).70,71 Alternatively, cancer may escape eradication at the later stages of the immune cycle, even after a vigorous effector T-cell response, by losing the ability to be destroyed (for example, by mutation of CASP8).72 The cancer microenvironment is characterised by hypoxia and decreased availability of nutrients required for energy and cell structure maintenance, including glucose, lipids, and amino acids.73,74 Anaerobic metabolism in the presence of oxygen (Warburg effect), a hallmark of cancer, leads to the production of large quantities of lactic acid that impair the function of immune cells. These metabolic changes lead to reprogramming of both the cancer cells and the immune cells in their microenvironment, resulting in a blunted immune response to the cancer and suppression of the effector CD8 T cells. Macrophage and myeloid cell differentiation is shifted to the immunosuppression type. Multiple therapeutic strategies have been proposed to overcome these obstacles.73,75
DEFINING PATIENT COHORTS: WHO BENEFITS?
The clinical experience with ICI has revealed that these drugs do not work for everyone; there are responders and non-responders, and only a minority of patients benefit.76 ICI work in defined cohorts of patients, relating to levels of expression of the PD-1/PD-L1 axis, expression of mutated genes that lend to nascent responses, and those that have tumour-infiltrating lymphocytes and other immune cells (i.e., hot tumours). This contributes to complexity in determining patient cohorts for vaccine trials, which requires consideration of the vaccine therapeutic mechanism to enhance the immune response in hot or inflamed tumours (characterised by tumour-infiltrating lymphocytes), or alter the immune response in cold tumours to make them ‘hot’. In addition, the PD-1/PD-L1 axis has the potential to be upregulated by transcriptional regulators that are yet to be defined but potentially associated with a cancer vaccine response. Some vaccines under consideration might have been dismissed because they upregulated transcriptional regulators known to shut down the immune response.
In the current era of immunotherapy, with the lack of definitive biomarkers, evaluation of tumours based on both their immune phenotype and genomic mutation profile may help determine which patients have a higher likelihood of responding to immunotherapies. Clinically, tumour burden reveals patient cohorts associated with therapeutic efficacy for cancer vaccines. Passively administered antibodies have been found to eliminate circulating tumour cells and systemic or intraperitoneal micrometastases in a variety of preclinical models; antibody-inducing vaccines may be beneficial in the adjuvant setting. Minimal residual disease is an indication for effective use of both monoclonal antibodies77 and for cancer vaccines.78 The results of the Keynote 522 Phase III clinical trial,79 comparing chemotherapy with pembrolizumab or placebo, revealed greater benefit in advanced stage of breast cancer than in early stage disease.
Insights and strategies from the immune foundation of ICI can be applied to the design and application of cancer vaccines, particularly to overcome the low antigenicity and heterogeneity of tumour-specific antigens. These include: 1) targeting multiple immunogenic antigens through polyvalent formulations;80-82 2) targeting a high fraction of tumour cells bearing each antigen, by considering the clonal nature of an antigen;83 and 3) deriving cancer vaccines from the most immunogenic clonal antigen-loaded patients.84The majority of patients are not responsive to ICI because of the lack of tumour-specific effector cells. Consequently, cancer vaccines may be a means to elicit diverse antigen-specific effector cells.85
Different measures of antigen-specific tolerance or regulation may help predict immunological outcome from vaccination.86 Santegoets et al.87 demonstrated prolonged overall survival following treatment with a cancer vaccine (GVAX) in combination with ipilimumab in patients with advanced prostate cancer who had either: high pre-treatment frequencies of CD4+ CTLA-4+, CD4+ PD-1+, or differentiated (non-naïve) CD8+ T cells; or low pre-treatment frequencies of regulatory T cells or differentiated CD4+ T cells. These parameters suggest a highly immunocompetent patient. Such findings suggest that the identification of predictive biomarkers associated with long-term immune outcome could be beneficial for identifying patients most likely to benefit from antitumour vaccines. One measure of immunocompetency, for consideration as an inclusion criterion for cohort recruitment, is delayed-type hypersensitivity to recall antigens.17 However, it has been suggested that this does not accurately reflect immune competence in patients with advanced- stage breast cancer, as research has demonstrated that patients who failed responses to recall antigens could still mount tumour-specific T-cell responses to a tumour antigen upon vaccination.88
Blank et al.89 suggested the integration of all the parameters involved in the immune response into one dynamic framework; they called it the ‘cancer immunogram.’90 Seven variables are included in this model: tumour foreignness, the patient’s general immune status, immune cell infiltration, checkpoints, soluble inhibitors, inhibitory tumour metabolism, and tumour sensitivity to immune effectors.
CONCLUSION
The ultimate goal of immunotherapy is to establish a durable population of highly active, tumour-specific responses that can lyse tumour cells and eradicate cancers. Evidence from various clinical trials that reflect the biology of immune response and cancer targeting lends to our understanding that cancer immunotherapy is a multifaceted strategy and that a single treatment modality will not suffice. The discovery of immune checkpoints and the success of their inhibitors has led to detailed investigation of the complicated interactions between different components of the immune system and microenvironment involved in the anticancer response.91 A plethora of co-stimulatory pathways have been identified, with some now the subject of intense investigation to assess the benefit of their activation for augmenting the anticancer immune response. Other inhibitory pathways were identified and are being explored to assess their role in different cancers. This line of research has revealed the complexity of the immune landscape.
CTLA-4 and PD-1/PD-L1 appear to be the predominant immune checkpoints, but they are not the only ones. Different tumours may preferentially utilise particular inhibitory pathways. Eliciting an immune response through a tumour vaccine may also trigger these specific inhibitory pathways. Research understanding at this point assumes that vaccines that lead to the release of high concentrations of INF-γ are likely to induce the overexpression of PD-L1 on tumour cells, and may benefit from the combination of the vaccine with PD-1/PD-L1 inhibitors. If a vaccine were to increase the expression of GAL9/Tim392 or GITRL/GITR,93 in addition to or instead of the PD-1/PD-L1 pathway, PD-1/PD-L1 inhibitors alone would be of limited use, as targeting the specific pathways triggered by the vaccine would be required. ICI rely on a primed nascent response. Cancer vaccines can provide priming and boosting of nascent responses but require ICI both to enhance a response, in the case of CTL-4 therapy, and to block tumour suppression, as in the case of the PD-1/PD-L1 axis.
Tumours are heterogeneous in their antigenic make-up, and different antigens of the same tumour may have different immunogenicity. Some cancers succeed in evading the immune system by decreasing their foreignness; others lose their expression of MHC-I and become invisible to the immune system. The metabolic microenvironment of the tumour favours the reprogramming of immune cells to Type 2 responses. Tumours differ in their ways of shutting off the immune system using different immune checkpoints. Hosts are also heterogeneous in their ability to mount an immune response to tumour cells. Vaccines that utilise tumour antigens could potentially induce similar responses to their respective tumours and may trigger different inhibitory mechanisms.
There is insufficient clinical data to reveal a breakthrough in cancer vaccines, but a better understanding of the tumour microenvironment allows for consideration of new combinations. Current understanding has determined that certain vaccines increase the release of IFN-γ, which in turn increases the expression of PD-L1 on the tumour, leading to immune suppression that can be overcome with the use of ICI. Questions remain concerning the timing of treatments, adjuvants, immunisation routes, optimal immunogenic vaccines, tumour remodelling, and the cohort these combinations should be tested in.
A rational approach to the development of vaccine-ICI combinations would require detailed definition of the tumour antigenic immunogenicity, immunogenic heterogeneity, and the inhibitory mechanisms that the tumour uses to suppress the immune system; in effect, a systems-based immunology/vaccinology approach is needed. With various vaccine modalities and combinations to alter the cancer microenvironment and the immune response under research, personalised immunotherapy could be a reality in the near future.






