
Abstract
In patients with haematologic malignancies such as lymphoma and leukaemia, significant increases in survival have been seen, due to advances in treatment strategies. However, long-term survivors of these cancers still remain at a higher risk of developing second cancers during their lifetime, especially in the fourth decade. It is well known that immunodeficiency after radiotherapy, chemotherapy, or haematopoietic stem cell transplantation impairs the antitumour activity of the innate and adaptive immune systems, predisposing the immune system to fail to detect and eliminate newly mutated cells. Literature shows evidence of various malignancies in these survivors, including sarcomas; breast cancers; mesothelioma; and solid secondary malignancies like breast cancer, thyroid cancer, and bone or soft tissue sarcoma.
Here, the author reports a case of development of Ewing’s sarcoma of the lung in an adult who is a long-term survivor of Hodgkin’s lymphoma.
INTRODUCTION
Hodgkin’s lymphoma (HL), first described by Sir Thomas Hodgkin in 1832, has evolved greatly in regard to its treatment. HL is a highly curable disease with a 5-year survival rate of approximately 88.3% overall, and more than 92.2% for localised disease.1 Due to the advances in diagnosis and treatment, there has been an increase in the number of survivors of HL. Most live for decades after their initial diagnosis and treatment. However, survivors of HL are at increased risk for late effects, which may manifest as subsequent primary cancers, cardiovascular disease, pulmonary toxicity, or endocrine dysfunction.2 These treatment effects can occur as late as the fourth decade after initial treatment.3
Ewing’s sarcoma (ES), a malignancy of adolescents and young adults, has an incidence of approximately one in 1 million, and is characterised by the proliferation of small round cells, expression of cluster of differentiation (CD) 99 in a characteristic membranous pattern, and non-random chromosomal translocations between the EWS gene on chromosome 22q12 and an ETS (E26 transformation-specific)family gene.4 It is the second most commonly occurring sarcoma, arising from bone or soft tissue in children. Few case studies have been published on ES occurring secondary to cancer therapy, including multimodal therapy for haematologic malignancies.5
Here, the author reports a case of development of ES of the lung in an adult who is a long-term survivor of childhood HL.
CASE REPORT
A 28-year-old male non-smoker was admitted with complaints of recurrent haemoptysis and right side diffuse, dull aching chest pain. They had a known case of HL diagnosed at the age of 13 years, for which he received six chemotherapy cycles of adriamycin, bleomycin, vincristine, and doxorubicin, and no radiotherapy. The patient was kept under follow-up. On further evaluation, the patient also gave a history of recurrent cold and cough associated with occasional dyspnoea since childhood. The patient had no history of recent fever, weight loss, or loss of appetite. Chest X-ray posteroanterior view was suggestive of right middle lobe collapse (Figure 1A). Lung function showed severe obstruction with significant reversibility. An empirical diagnosis of uncontrolled bronchial asthma was made. The patient was further evaluated for allergic bronchopulmonary aspergillosis. Serum total IgE (940 IU) and specific IgE (0.34 IU) against Aspergillus fumigatus were raised. Allergic bronchopulmonary aspergillosis antifungals (voriconazole and caspofungin), oral corticosteroids, and inhaler therapy were prescribed. However, during follow-up, the patient continued to have haemoptysis, and repeat chest X-rays showed complete collapse of the right lung (Figure 1B). CT pulmonary angiogram was suggestive of a fairly-defined, poorly-enhanced lobulated soft tissue density in the right hilar lesion encasing the right bronchus and pulmonary artery, and causing narrowing of the right upper and middle lobe bronchi, and complete occlusion of lower bronchus with multiple patchy ground-glass opacities, likely to be right lower lobe complete collapse with alveolar haemorrhages (Figure 2).
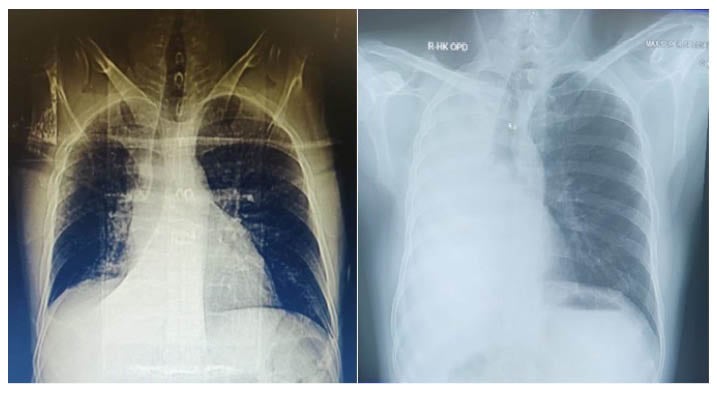
Figure 1: Chest X-ray of a patient with uncontrolled bronchial asthma, who had been diagnosed Hodgkin’s lymphoma.
A) Chest X-ray at time of presentation showing right middle lobe collapse. B) Follow-up chest X-ray showing complete collapse of right middle side with mediastinal shift on same side.
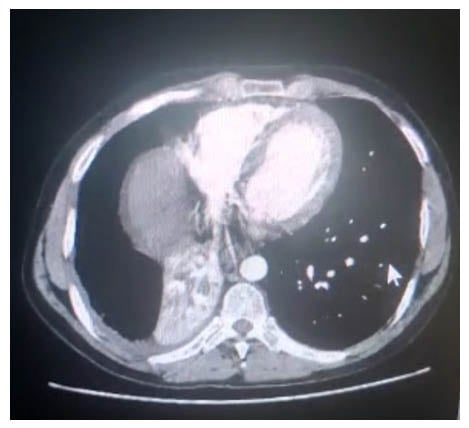
Figure 2: Mediastinal window of chest CT showing lobulated soft tissue density in the right hilar lesion.
The patient underwent fiberoptic bronchoscopy, which showed an intraluminal mass causing complete obliteration of the right main bronchus up to the carina. Bronchoalveolar lavage for malignant cells was inconclusive. Endobronchial biopsy showed infiltration of sheet of polygonal/round tumour cells in respiratory epithelium submucosa, with tumour cells having scant cytoplasm, a high nucleocytoplasmic ratio, and coarse chromatin with inconspicuous eosinophilic nucleoli. Endobronchial ultrasound-guided transbronchial needle aspiration showed singly scattered and clusters of markedly pleomorphic tumour cells with coarse chromatin, conspicuous nucleoli, and a scant-to-moderate amount of cytoplasm, which was positive for malignant cells (Figure 3A). On immunohistochemistry, it was positive for vimentin and CD99. They showed nuclear transducing-like enhancement of split expression and FLT-1 positivity, and were negative for cytokeratin 7, cytokeratin 5/6, epithelial membrane antigen, leukocyte common antigen, PO, thyroid-specific transcription factor-1, synaptophysin, chromogranin, CD138, S-100, con, CD30, CD3, CD34, smooth muscle actin, and desmin. The Ki-67 proliferative index was approximately 10%, favouring a diagnosis of malignant round cell tumour, likely to be primitive neuroectodermal tumour. Further, in cytogenetics, fluorescence in situ hybridisation for EWSR1 (22q12.2) gene rearrangement was positive in 68% of tumour cells (Figure 3B), whereas there was no evidence of translocation (X;18)(p11.2;q11.2)/SS18 gene rearrangement (Figure 3C). A diagnosis of ES of the lung was made, and the patient planned for chemotherapy. However, the patient defaulted for further evaluation, and received alternative medicine for a period. The patient again presented with persistent cough and dyspnoea, requiring intensive care unit admission. A PET-CT scan showed an ill-defined heterogeneously hypermetabolic pulmonary mass arising from the right lung field, with associated hypermetabolic collapse. Presence of hypermetabolic pleural, pulmonary, lymphatic, and skeletal metastasis was also seen, confirming diagnosis of Stage IV ES. Bone marrow aspiration and biopsy were performed as a staging workup, which showed trilineage haematopoiesis with normocellular marrow.
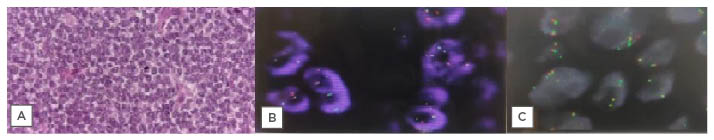
Figure 3: A series of tests leading to the diagnosis of Ewing’s sarcoma of the lung.
A) Histopathology of EBUS-TBNA sample. B) FISH showing positive tumour cell for EWSRI gene rearrangement. C) Showing no evidence of translocation SS18 gene rearrangement.
EBUS-TBNA: endobronchial ultrasound-guided transbronchial needle aspiration; FISH: fluorescence in situ hybridisation.
The patient was then treated with a regimen of vincristine, adriamycin, and cyclophosphamide (VAC). The patient had a symptomatic response to the first cycle of VAC, so another cycle was repeated. After the second cycle, the regimen was changed because the patient had already received 420 mg/m2 of adriamycin (300 mg in six cycles of adriamycin, bleomycin, vincristine, and doxorubicin given during HL treatment, and 120 mg in two cycles of VAC). A new regimen containing vincristine, ifosfamide, and etoposide was given for two cycles, followed by two further cycles of irinotecan and temozolomide. The patient tolerated this therapy well; however, repeat PET-CT scans showed disease progression, with an increase in the size and metabolic activity of the primary and metastatic lesions, and with development of new nodal and soft tissue metastasis.
The patient was then treated with three cycles of the next line of chemotherapy, gemcitabine and docetaxel, but the disease progressed both radiologically and clinically. The patient was advised to undergo palliative radiotherapy in view of the recurrent cough and breathing difficulty, but refused to undergo radiotherapy, and was lost to follow-up.
DISCUSSION
There has been a significant increase in the survival of patients with haematologic malignancies as a result of advances in treatment strategies, including those for indolent and aggressive forms of lymphoma and leukaemia.6,7, Long-term survivors of these cancers still remain at a higher risk of developing second cancer during their lifetime because of multifactorial causes.
Risk Factors Responsible for Second Cancer
There are numerous risk factors, such as age at treatment,8 sex, phenotype of the first cancer, treatment modalities, radiation dose,9 and chemotherapy10,11 of the first cancer. Several treatment modalities for newly diagnosed malignancies have been implicated as risk factors for second malignancy, including various chemotherapeutic agents, radiotherapy, targeted therapy with monoclonal antibodies, and stem cell transplantation. Treatments are decided according to the phenotype, the line of differentiation, biological markers, and extent of the disease. Patients who received radiation therapy for first cancer are more prone to develop sarcomas, breast cancers, and mesothelioma compared with people who did not receive it. The most frequently observed solid secondary malignancies are breast cancer, thyroid cancer, and bone or soft tissue sarcomas.12 With extended follow-up of cohorts of young survivors of HL, increased risks of common adult carcinomas, including colorectal, lung, and gastric types, have emerged, and these cancers are being diagnosed at younger ages than observed in the general population.13
The use of treatment regimens based on a reduction in the field and dose of radiotherapy and alkylating chemotherapy has been introduced in order to reduce rates of long-term complications, while maintaining a high cure rate. Despite such modifications, a recent study from the Netherlands showed that this has not affected the risk of second cancers in individuals with HL.3 Applebaum et al.14 found that 12.1% of patients developed ES at the site of irradiation for the initial cancer.
Pathogenesis
It is well known that immunodeficiency after radiotherapy, chemotherapy, or haematopoietic stem cell transplantation impairs the antitumour activity of the innate and adaptive immune systems.11 This predisposes the immune system to fail to detect and eliminate newly mutated cells carrying the ES translocation, allowing for the initiation of secondary ES.
The pathogenesis of ES is strongly linked to the presence of a translocation between the EWSR1 gene and ETS gene family members. In 80% of cases, the partner gene is FLI1, with translocation (11;22)(q24;q12) leading to EWS–FLI1.15 ES involves the expression of a germline predisposition syndrome, accounting for the strong predilection of ES to occur in young patients, similar to other genetically linked tumours.
Histology
Histologically, the tumour consists of a proliferation of small round cells with scanty and clear cytoplasm, round to oval nuclei, finely granular chromatin, and inconspicuous nucleoli. It is periodic acid–Schiff-positive due to the presence of cytoplasmic glycogen. Histologic differential diagnoses include small cell carcinoma, malignant lymphoma, alveolar rhabdomyosarcoma, and neuroblastoma. Tumours have a strong reactivity to CD99/MIC-2 and vimentin. In some cases, they may be positive for markers of neural differentiation, such as S100, neuron-specific enolase, and cytokeratins (in 20% of cases). Demonstration of translocation (11;22)(q24;q12) by fluorescence in situ hybridisation and/or reverse transcription-PCR is used to support the diagnosis.16
Treatment Options
There are only a few reports on ES as a secondary malignancy in the literature, mostly comprising single case reports or short series describing ES following treatment for unrelated tumours. As such, there are no defined guidelines or directions for managing such situations; hence, more experience is needed. The treatment of ES is aggressive, with the most effective approach being surgical resection with combination chemotherapy and/or high-dose radiation therapy. Prognosis is mainly related to the ability to achieve disease-free surgical margins, and the extent of anatomical spread to surrounding structures such as bone, pleura, and the epidural space.17,18 The standard first-line treatment for patients with these tumours has been based on five drugs mainly included vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide. Pazopanib efficacy in pulmonary ES was reported, but it is not considered a standard therapy.19
CONCLUSION
The author has described an extremely rare case of pulmonary ES in a case of treated HL. Though rare, it should be considered in the differential diagnosis of patients with a history of HL. It is important for physicians and oncologists to follow-up patients with HL in the long term for accurate monitoring and quantification of the associated risks of second cancers and other late effects.






