Abstract
Pseudomyogenic hemangioendothelioma is an uncommon vascular neoplasm that has recently been identified as a distinct entity. Despite being classified as a malignant tumour and its often-worrisome clinical presentation, the progression typically involves relapses after surgery and a rather infrequent potential for metastasis.
The authors present the clinical case of a pseudomyogenic hemangioendothelioma of the lower limb, its evolution to lymph node involvement and distant metastasis, and the effectiveness of vinorelbine in its management. Through this case report, the authors underscore the significance of a precise histological and immunohistochemical assessment considering the usual misdiagnosis as sarcoma or metaplastic carcinoma. They also emphasise the significance of observing the evolutionary aspects of this indolent tumour while outlining the therapeutic strategy and systemic therapies’ sequences to enhance the quality of life for these long survivors.
Key Points
1. Pseudomyogenic hemangioendothelioma is a scarce tumour with distinct clinicopathological features that raise diagnostic and therapeutic challenges.2. Advances in molecular genetics and mostly immunohistochemistry have allowed better diagnostic criteria and hence an earlier diagnosis.
3. A series of individual chemotherapy treatments over time appears to be the most effective approach for metastatic or recurrent pseudomyogenic hemangioendothelioma.
INTRODUCTION
Pseudomyogenic hemangioendothelioma (PMH) is an uncommon vascular tumour with challenges often in its diagnosis and treatment. It is classified as an endothelial neoplasm that rarely metastasises and typically exhibits a slow-growing, indolent nature. No universally accepted standard of care has been defined for such cases; nevertheless, the literature suggests a wide range of treatment options varying from local control surgeries and cryotherapy all the way to systemic therapies such as tyrosine kinase inhibitors and bone resorptive agents. The aim of this case report is to describe a frequent instance of PMH and to examine its clinical and pathological features, treatment options, evolutionary aspects, and prognosis.
CASE REPORT
The authors report the case of a 66-year-old man with no notable prior medical history who presented with a growing lump and right knee pain for over 6 months. Physical examination revealed a 30 mm multinodular mass of the crural stump of the right knee.
A first excisional surgery was performed, and no specimen was provided for pathological analysis at that time. This was followed by a second surgery for local recurrence within a year.
The surgical wound exhibited impaired healing, and the patient underwent an amputation of the lower limb. He was then eventually referred to the authors’ department for continued management and care.
The histologic examination each time revealed a tumour proliferation made of sheets and short fascicles. Tumour cells were spindle to epithelioid with vesicular nuclei and abundant eosinophilic cytoplasm. Hemangiopericytoma-like vasculature was observed (Figure 1). Immunohistochemical (IHC) study showed strong expression of ERG and focal expression of AE1-AE3. Tumour cells were negative for h-caldesmon, smooth muscle actin, desmin, epithelial membrane antigen (EMA), CD34, and S100 protein (Figure 2). Based on all these results, a diagnosis of PMH was made.
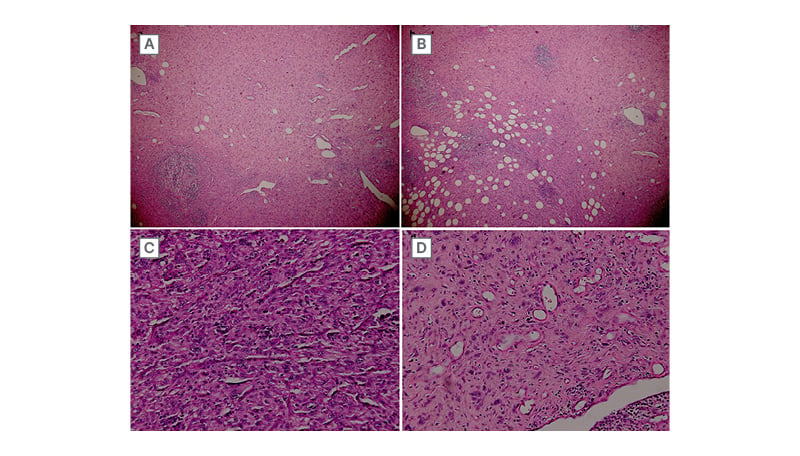
Figure 1: Representative micrographs of the tumour.
Tumour cells are arranged in sheets or in short fascicles. A) Hemangiopericytoma-like vasculature is observed. B) Tumour infiltrates surrounding adipose tissue. C and D) Tumour cells are plump spindle to epithelioid with vesicular nuclei and abundant eosinophilic cytoplasm.
(haematoxylin-eosin; A: x40, B: x100, C and D: x200).
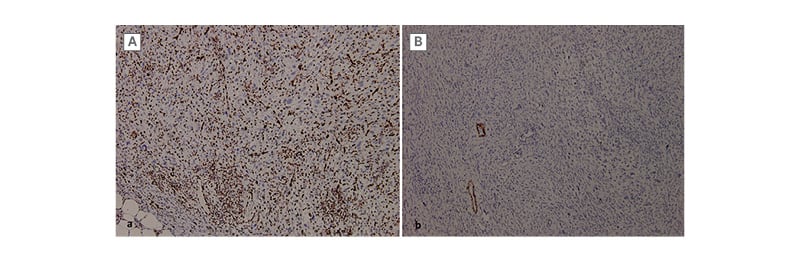
Figure 2: Immunohistochemical profile of the lesion.
A) The tumour cells are positive for ERG. B) They are negative for h-caldesmon (with blood vessels as
positive control).
An MRI of the right thigh showed a subcutaneous mass of right crural bump with lobulated contours, heterogeneous tumour-like tissue. Through the MRI, a second similar mass on the track of the sciatic nerve was identified (Figure 3). CT scan showed a subdiaphragmatic lymph node associated with secondary pulmonary lesions. An ultrasound biopsy was performed to confirm the nature of the mass. The biopsy and IHC appearance were in favour of PMH.
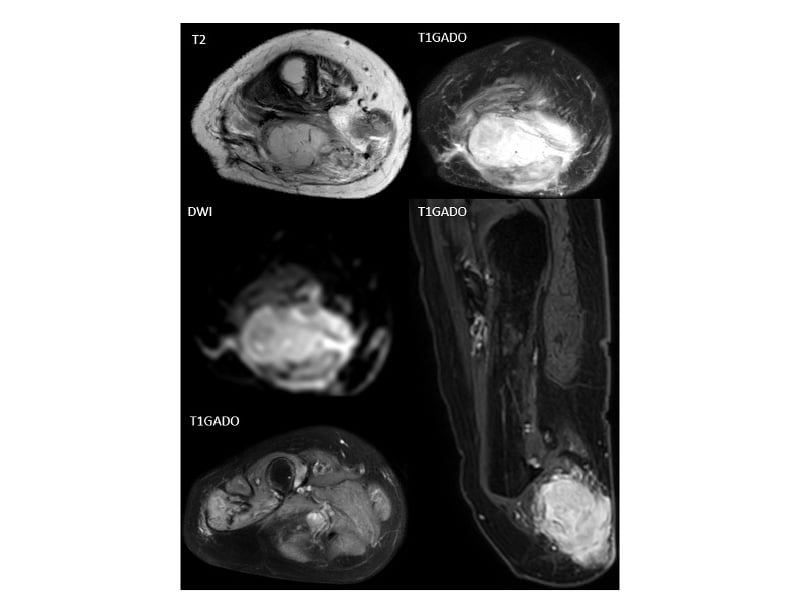
Figure 3: MRI of the right amputated leg.
MRI of the right amputated leg shows a tissue mass in the posterior region of the right thigh located opposite the
femoral amputation stump, with an isointense signal on T1 relatively to the adjacent muscles and a high signal intensity on T2 with restriction in diffusion, and an intense heterogeneous enhancement after gadolinium injection. Locally, the mass invaded the muscular fascia with extension to the facing subcutaneous soft tissues. MRI identified a second mass with identical characteristics to the previous one located on the distal portion of the sciatic nerve (yellow square).
Given the multifocal nature of the disease, the patient was deemed unsuitable for surgical resection and was started on a gemcitabine 900 mg/m2 and docetaxel 75 mg/m2 regimen with no notable adverse events.
The evaluation after the third session showed clinical and radiological progression, and vinorelbine as a second line was recommended by an expert in the European Soft Tissue and Bone Sarcoma Group (STBSG). The patient showed good clinical response to treatment, demonstrating a volume reduction of primary and secondary sites and a stable disease within 8 months of follow-up.
The patient is currently on treatment with a good overall performance status ranging from 2 to 1. Ongoing monitoring for disease progression or potential complications is being maintained.
DISCUSSION
PMH, alternatively referred to as epithelioid sarcoma-like hemangioendothelioma, which exhibits morphological characteristics resembling those of a myoid neoplasm or an epithelioid carcinoma, is now regarded as a unique endothelial neoplasm with minimal propensity for metastasis.1
In a series of 50 cases of PMH, the majority of patients (82%) were male and under 40 years of age. Lower extremity involvement was prevalent (54%), with a predominance of soft-tissue lesions. Multifocal lesions were present in 66% of the cases.2
Diagnosis of PMH is usually based on clinical presentation, histological examination, and verifying the manifestation of both keratins and endothelial markers via IHC analysis.
Most patients remain asymptomatic up to 10 years before the diagnosis,3 while 60% of patients with PMH will experience local recurrence,2 which indicates its locally aggressive but relatively indolent profile.
PMH typically presents as lobular, clearly demarcated lesions. Cross-sectional imaging modalities are vital for diagnosing and locating PMH, given its manifestation across diverse anatomical sites.
On conventional radiographic imaging, PMH of bone presents as osteolytic, lobular, and proliferative lesions, with well-defined borders and a sclerotic rim, but without periosteal reaction. These lesions can lead to cortical destruction and pathological fractures. CT imaging shows similar characteristics, while soft tissue lesions, as exemplified by the case of the authors’ patient, manifest as indeterminate, lobular formations with well-defined demarcations.
On MRI, PMH-indicative lesions will usually exhibit low-to-intermediate signal intensity on T1-weighted sequences and will appear hyperintense on T2-weighted sequences. T1 fat-saturated post-gadolinium enhanced MRI aids in the recognition of intramuscular localisations, which appear as clearly delineated, hyperintense foci with notable contrast enhancement.
A comprehensive whole-body PET-CT scan is advised owing to the disease’s prevalent diffused manifestation.
Although whole-body MRI has great sensitivity, PET-CT is preferred for detecting metabolic activity in lesions, showing high fluorodeoxyglucose uptake. To date, all medical practitioners have employed PET-CT for lesion localisation as opposed to utilising whole-body MRI.4-5
Immunohistochemically, these tumours show widespread expression of cytokeratin (AE1/AE3), primarily in focal areas.6 In some instances, there is occasional staining for epithelial membrane antigen and smooth muscle actin.7 In this study, the tumour cells were negative for anti-PS100, desmin, EMA, and CD34, but all showed focal expression of cytokeratin (AE1-AE3), which was consistent with the literature.
The main distinguishing characteristics of PMH in contrast to other forms of hemangioendothelioma are the variability in CD31 expression, the nonexistence of CD34 expression, and the absence of discernible histological indicators of endothelial differentiation.3
The primary differential diagnosis of PMH includes other vascular tumours (epithelioid haemangioma, and epithelioid hemangioendothelioma), epithelioid sarcoma, and metaplasic carcinoma with sarcomatoid features.8
Surgical removal with clear margins remains the primary treatment option for PMH; however, achieving negative margins can be challenging due to the tumour’s infiltrative nature and multifocal growth pattern.
Due to the rare occurrence of PMH, clinical investigations are absent. However, a range of systemic therapeutic modalities have been documented in clinical case studies.
Given the recurrent local relapses and disseminated distribution of PMH, a series of individual chemotherapy treatments over time appears to be the most effective approach, such as weekly gemcitabine, doxorubicin, oral vinorelbine, and oral cyclophosphamide, which has ensured satisfactory local tumour control.
Sirolimus and everolimus, known as mTOR inhibitors, have shown significant efficacy, especially for tumours where the DNA sequencing revealed a TSC1 mutation. While mTOR inhibitors are not currently established as definitive curative treatments, they can be utilised for long-term and maintenance therapy with minimal side effects.9
Patients with extensive disease under tyrosine kinase inhibitors such as pazopanib have demonstrated a positive clinical and radiological response, with sustained disease regression observable within a 6-month follow-up period.10
The current National Comprehensive Cancer Network (NCCN) guidelines recommend the use of pazopanib (mostly for patients unfit for intravenous systemic therapy or patients who are not candidates for anthracycline-based regimens), sirolimus, and anthracycline-based regimens as first-line treatment for advanced or metastatic disease.1
Brance ML et al.11 reported a favourable response to pamidronate (90 mg/monthly) for a locally advanced PMH with osseous and soft tissue dissemination, and a PET-CT that showed no metabolically active site in a 3-year follow-up.
In a comparable manner, the two case reports have shown encouraging results where 4 weekly doses of denosumab have resulted in stable disease for up to 4 years and mostly an alleviation of pain even in multifocal osseous lesions.6-12
CONCLUSION
PMH is a scarce tumour with distinct clinicopathological features that raise diagnostic and therapeutic challenges. Advances in molecular genetics and immunohistochemistry have improved the understanding of its pathogenesis and diagnostic criteria. This advancement has allowed for a reduction in the aggressive surgical approach, which typically leans towards amputation, especially when the tumour affects limbs.
Further studies are needed to elucidate the underlying mechanisms driving its aggressive behaviour and to identify a more effective standard of care for this challenging entity.
PATIENT PERSPECTIVE
“After my diagnosis, I experienced significant anxiety; however, the thorough explanations from the healthcare team were instrumental in alleviating my concerns. After the second line of treatment, I noticed substantial improvements in my symptoms, mostly in the alleviation of pain, which also improved my emotional health.”






