
Abstract
Metastatic colorectal cancer (mCRC) is a markedly heterogeneous disease, which portends a poor prognosis, with an estimated 5-year overall survival rate of approximately 15%. The standard of care of systemic therapy remains fluoropyrimidine-based chemotherapy, with modest results, despite improvements with the combination with anti-angiogenics and anti-epidermal growth factor receptor therapy.
Significant advances in cancer therapy have been observed in the past two decades. The enhanced appreciation of molecular biology in oncology has allowed for the identification of specific molecular subtypes and novel therapeutic targets. Nevertheless, meaningful precision-based advancements in the therapeutic options for mCRC have been challenging and slow to realisation. Comprehensive molecular profiling and circulating tumour DNA highlight a heterogeneous disease at the genomic, epigenomic, and transcriptomic levels, and with a low frequency of actionable alterations.
In the present review, the authors describe the current and emerging predictive biomarkers in mCRC, as well as present landmark clinical trials that have allowed for evolving precision in the therapeutic management. The understanding of the benefit of immune checkpoint inhibitors in patients with high microsatellite instability cancer and in those with POLE mutations or high tumour mutational burden, the combination of BRAF with epidermal growth factor receptor inhibition in BRAF V600-mutated patients, the use of allele-specific KRAS G12C inhibitors, the promising findings of dual anti-HER2 therapy in HER2-positive mCRC, and the possibility to offer targeted therapy for patients harbouring gene fusions NTRK/ALK/ROS1 have ushered in a new era of precision oncology for mCRC, providing personalised treatments and sustaining hope for patients affected by this challenging disease.
INTRODUCTION
Colorectal cancer (CRC) is the second most common cause of cancer-related death in the USA.1 Despite improvements in screening rates and in the overall survival (OS) of patients with localised and advanced disease over the past few decades, the 5-year OS of patients with metastatic disease is still extremely poor and estimated to be approximately 15%.2,3
Significant advances in cancer therapy have been observed in the past two decades. The enhanced appreciation of molecular biology in oncology has allowed for the identification of specific molecular subtypes and novel therapeutic targets. This era of precision oncology allows for the development of biomarker-guided therapeutics and has markedly transformed the landscape of cancer treatment. Precision medicine represents a paradigm shift in oncology, moving from a histology-based chemotherapy to include genome-specific targeted therapy, which has promoted ongoing discovery for novel biomarkers in all malignancies.
Nevertheless, the emergence of precision oncology has drastically improved the management of CRC. Comprehensive molecular profiling confirms a markedly heterogeneous disease at the genomic, epigenomic, and transcriptomic levels, but currently with low frequency of actionable alterations. For more than a decade, personalised therapy in CRC was restricted to the identification of RAS mutations, which are predictive markers of resistance to anti-epidermal growth factor receptor (EGFR) monoclonal antibodies. Only recently, clinical trials addressing novel genome-guided personalised therapies in specific molecular subtypes of CRC have been successfully completed, expanding the clinical relevance of precision oncology in CRC.
In this review, the authors describe the current and emerging predictive biomarkers in metastatic CRC (mCRC), as well as present landmark clinical trials that have allowed for evolving precision in the management of this heterogenous disease.
ANTI-EPIDERMAL GROWTH FACTOR RECEPTOR MONOCLONAL ANTIBODIES IN METASTATIC COLORECTAL CANCER IN THE PAST TWO DECADES: PRECISION THAT NEEDS FURTHER REFINEMENT
For almost two decades, the clinical applicability of precision in mCRC has been limited to the use of the anti-EGFR monoclonal antibodies cetuximab and panitumumab for RAS wild-type disease. The knowledge accumulated over the past 20 years has demonstrated that the benefit offered by these monoclonal antibodies is restricted to a smaller subset of mCRC than initially proposed (Figure 1). Pure predictive biomarkers reflecting patient specific sensitivity to anti-EGFR monoclonal antibodies remains a developing area in mCRC. Interestingly, evidence accumulated over the past few years suggest that not only expanded RAS mutations such as NRAS and HRAS may confer additional resistance to cetuximab or panitumumab but also BRAF, PI3KCA, HER2, MET, PTEN, and AKT1 abnormalities, as well as NTRK/ROS1/ALK/RET rearrangements.4-9 Furthermore, recent studies have consistently demonstrated that patients with right-sided tumours derive lower, if any, benefit from that therapy.10,11
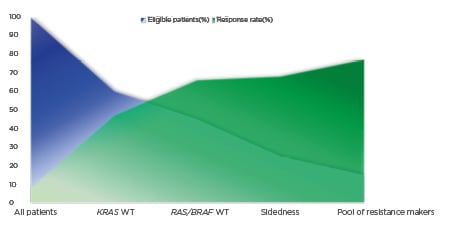
Figure 1: Negative hyper-selection of patients with metastatic colorectal cancer to anti-epidermal growth factor receptor monoclonal antibodies.
The graph shows the ascending response rate (y-axis, in green) of anti-EGFR therapy in mCRC based on patient selection by biomarkers (RAS and BRAF), sidedness, and a pool of resistance markers such as BRAF, PI3KCA, HER2, MET, PTEN, and AKT1 abnormalities, as well as NTRK/ROS1/ALK/RET rearrangements, which lead to a descending rate of eligible patients for targeted therapy (x axis, in blue). Since there are no predictive biomarkers of sensitivity for anti-EGFR therapy, the patient selection based on predictive markers of resistance may be denominated as negative hyper-selection.
EGRF: epidermal growth factor receptor; mCRC: metastatic colorectal cancer; WT: wild-type.
Hence, the estimated rate of patients with mCRC who are actually sensitive to cetuximab or panitumumab is lower than approximately 15%.9 The current recommendation to use anti-EGFR monoclonal antibodies in patients with left-sided tumours and RAS/BRAF wild-type status still is an incipient and imprecise clinical applicability of precision medicine in the systemic therapy of CRC. Therefore, the identification of refined biomarkers and novel targeted therapies are urgently needed (Figure 2; Table 1).
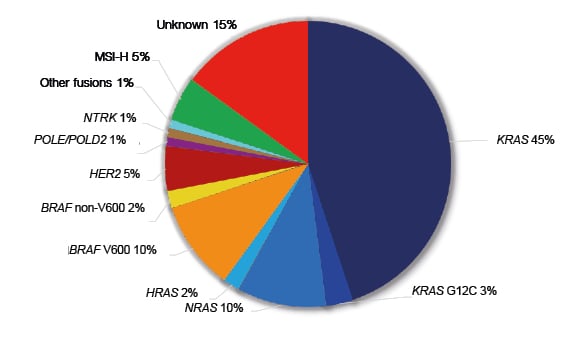
Figure 2: Molecular alterations with therapeutic implications in metastatic colorectal cancer.
MSI-H: high-frequency microsatellite instability.
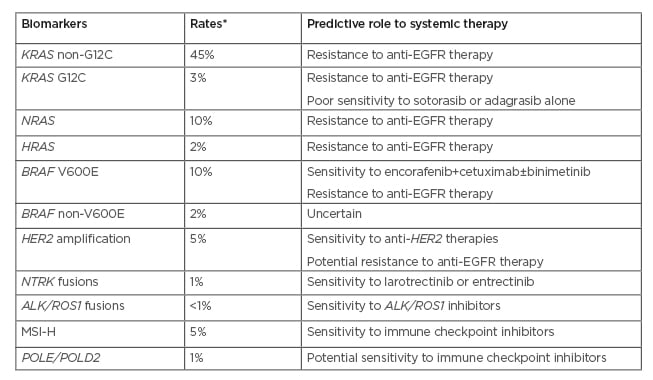
Table 1: Predictive biomarkers and targeted therapies in metastatic CRC.
*Approximate rates.
MSI-H: high-frequency microsatellite instability, EGFR: epidermal growth factor receptor.
NOVEL THERAPEUTIC TARGETS IN COLORECTAL CANCER
MSI-H CRC
It is estimated that approximately 5% of patients with mCRC harbour high-frequency microsatellite instability (MSI-H), which might originate from two mechanisms: somatic hypermethylation of the MLH1 gene promoter, commonly associated with BRAF V600E mutation; or point mutation of one of the mismatch repair genes, mainly MLH1 and MSH2.12,13 Patients with MSI-H CRC compose a subgroup with distinct molecular and clinical characteristics. Typically, they present a younger median age at diagnosis, with tumours predominantly located at the proximal colon, commonly with lymphocyte infiltration, and with a higher median number of tumour mutational burden (TMB).13 In addition, they have lower sensitivity to chemotherapy compared with patients classified as microsatellite stable (MSS), and, more importantly, they tend to be sensitive to immunotherapeutic approaches, such as immune checkpoint inhibitors (ICIs).
Encouraging data from Phase I and II clinical trials14-19 prompted the conception of the Phase III KEYNOTE-177 study, which compared the efficacy of standard chemotherapy (doublets plus anti-vascular endothelial growth factor or anti-EGFR) with pembrolizumab in 307 treatment-naïve patients with MSI-H mCRC.20 One of the primary endpoints, progression-free survival, was met: 8.2 months in the chemotherapy group versus 16.5 months in the immunotherapy group (hazard ratio [HR]: 0.60; 95% confidence interval [CI]: 0.45–0.80; p=0.0002). Likewise, the overall response rate (ORR) was statistically higher in the immunotherapy arm: 33.1% versus 43.8%.
Interestingly, 29.4% of the patients in the pembrolizumab group presented progressive disease compared with 12.3% in the chemotherapy group, predominantly in the first 4 months of treatment. Of the patients who presented objective response to immunotherapy, an impressive rate of 83% had ongoing responses at 24 months, compared with only 35% in the chemotherapy group. Another primary endpoint, OS, did not have mature data to be analysed.
MSI-H is not the only biomarker to explain sensitivity to ICIs. Patients with MSI-H who present with low TMB seem to have a lower probability to respond to immunotherapy.21 A high concordance rate is expected between MSI-H and TMB. Patients with abnormalities in DNA mismatch repair pathways, whether germline or somatic, tend to present higher number of nonsynonymous mutations, and thereby high TMB. In a study with 6,004 patients with mCRC, 5% were classified as MSI-H and 95% as MSS.22 The median TMB was significantly higher in the population with MSI-H: 46.8 mutations/Mb versus 3.6 mutations/Mb. The median TMB in the overall population was 4.5 mutations/Mb. Approximately 3% of patients with MSS were classified as high TMB, defined as ≥12 mutations/Mb. Variants in the mismatch repair genes, such as MLH1, MSH2, and MSH6, as well as in POLE, were significantly more common in this population of patients with high TMB and MSS relative to those with low TMB and MSS.22
POLE
POLE encodes the catalytic subunit of DNA polymerase ε, which acts in the replication of the DNA strand before cell division.23POLE proofreading is an essential step in the maintenance of the integrity of the genome, which is consistent with the finding of ultra-mutated tumours in the presence of pathogenic exonuclease domain mutations, with a mean tumour mutational burden >200 mutations/Mb.24,25POLE mutation is rarely found in malignancies, being identified in approximately 5–10% of endometrial cancer,26 1% of CRC,27 and less frequently in gastric and pancreatic cancers.28
POLE-mutated CRC portends a better prognosis. In a population of 6,517 patients with CRC, 1% (66 patients) harboured the mutation, which was associated with a reduced risk of recurrence and a superior OS in a population of patients with Stage II/III CRC.23 Patients with POLE-mutated CRC were younger at diagnosis, predominantly male, with a higher frequency of right-sided tumours, and with disease diagnosed at earlier stages compared with the wild-type counterparts.23,25,29 They also demonstrated increased CD8+ lymphocyte infiltration and expression of cytotoxic T-cell markers.23
This immunogenic subset of CRC has been demonstrated to be highly sensitive to the ICIs. Case reports with successful experiences in the treatment of metastatic CRC have been presented in the past few years.31,32 Since there is a U.S. Food and Drug Administration (FDA) approval for the use of pembrolizumab for patients with metastatic disease and TMB >10 mutations/Mb,32 the use of immunotherapy should be strongly considered in the treatment of patients with POLE-mutated mCRC. The probability to identify these mutations is higher in early-onset CRC compared to the late-onset.33
BRAF
BRAF V600E mutation is found in approximately 10% of patients with mCRC.34-37 These patients have a poorer prognosis compared with the wild-type counterparts, demonstrating lower sensitivity to the standard chemotherapeutic drugs used in CRC, with lower ORR, and shorter progression-free survival and OS.38-40 More commonly found in right-sided tumours, this mutation, similarly to RAS mutations, also denotes resistance to the anti-EGFR monoclonal antibodies.41,42
The success of BRAF inhibitors in the systemic therapy of BRAF-mutated melanoma prompted the evaluation of these drugs in mCRC. However, Phase I data addressing the efficacy of vemurafenib in patients with BRAF-mutated mCRC showed poor efficacy.43 Preclinical studies demonstrated that BRAF inhibition induced adaptive feedback reactivation of mitogen-activated protein kinase signalling, often mediated by EGFR activation, suggesting that the combination of BRAF inhibitor with an anti-EGFR monoclonal antibody might overcome this therapeutic resistance.44-46 Subsequently, a Phase IB study confirmed the hypothesis, demonstrating that the combination of vemurafenib, irinotecan, and cetuximab yielded 35% of ORR in a population of 19 patients with BRAF-mutated mCRC.47 Additionally, further work demonstrated the clinical activity of the combination of BRAF and EGFR inhibition with or without mitogen-activated protein kinase kinase (MEK) inhibition in BRAF-mutated mCRC.44
These promising findings elicited the conception of the Phase III BEACON study, a three-arm clinical trial that explored the combination of BRAF and EGFR inhibition with MEK inhibition. A total of 665 patients with BRAF V600E-mutated mCRC who had been submitted to at least one previous line of systemic therapy were randomised to one of three arms: the triplet-regimen composed of encorafenib plus binimetinib plus cetuximab; the doublet-regimen with encorafenib plus cetuximab; and the control arm with irinotecan-based regimens (folinic acid, fluorouracil, and irinotecan; or irinotecan) plus cetuximab.48 The primary end points were OS and ORR in the triplet-regimen arm compared to the control arm. Updated survival results showed a median OS of 9.3 months in the triplet arm versus 5.9 months in the control arm (HR: 0.60; 95% CI: 0.47–0.75).49 The ORR was 27%, 20%, and 2% in the triplet, doublet, and control arms, respectively. A comparison of the median OS in the doublet arm (9.3 months) with the control arm, a secondary endpoint, also favoured the BRAF inhibitor (HR: 0.61; 95% CI: 0.48–0.77). There was no statistically significant difference between the triplet and doublet arms in OS: 9.3 months in both groups (HR: 0.95; 95% CI: 0.74–1.21).49 Grade ≥3 adverse events were found in 66%, 58%, and 64% of the patients in the triplet, doublet, and control arms, respectively. Based on BEACON data, the FDA has approved the combination of encorafenib plus cetuximab for the treatment of patients with mCRC and a BRAF V600E mutation with at least one prior systemic therapy.50
The clinical relevance of non-V600 BRAF mutations has not yet been fully elucidated. These mutations have been found in approximately 2% of patients with mCRC, of which the D594N (Class III) and G469A (Class II) mutations seem to be the most frequent.51 The patients who harbour these atypical BRAF mutations seem to present similar prognosis compared to the wild-type counterparts. Unlike V600E, these atypical mutations are mostly identified in left-sided tumours, and younger male patients.51 In addition, most of them are MSS and RAS mutations are not mutually exclusive in this context, occurring in approximately one-third of non-V600 patients.51,52 The predictive value of these mutations for the deployment of anti-EGFR monoclonal antibodies is not yet entirely clear, and appears to differ according to the underlying BRAF class. Class II mutations appear to be resistant while Class III are sensitive, although with limited duration.53-55 Furthermore, non-V600 BRAF mutations might be involved in the development of adaptive resistance to EGFR inhibition.51 These distinct class-specific biochemical and functional properties highlight the importance to decipher the unique biology of atypical BRAF mutations in order to promote novel clinical trial design and ultimately offer effective therapeutic options for patients.
KRAS G12C
KRAS mutations are the most common activating genetic mutations in solid tumours, mainly in pancreatic cancer, non-small cell lung cancer, and CRC, where they are estimated to be found in approximately 45% of tumours.37,56,57 Right-sided tumours present a higher percentage of KRAS mutations compared with their left-sided counterparts, mainly in the cecum, where approximately 70% of the tumours harbour the mutation.56 For decades, KRAS mutations have not been deemed as actionable, but, together with other RAS mutations, they predict resistance to anti-EGFR monoclonal antibodies in CRC.58,59
Despite years of research focus, targeting KRAS has been an elusive goal in cancer therapy since the mutated protein has high affinity for guanosine triphosphate or guanosine diphosphate and has no binding pocket.60 In addition, inhibition of the downstream effectors in the mitogen-activated protein kinase pathway (BRAF-MEK-ERK) has proven ineffective in clinical trials.60
The codons 12 and 13 in exon 2 are the most commonly altered in KRAS mutations, occurring in approximately 30% and 10%, respectively, of the patients with mCRC.57 The amino acid changes p.G12D, p.G12V, and p.G13D are the most frequent of these codons in CRC, found in approximately 13%, 10%, and 9% of the patients, respectively.57 The oncoprotein KRAS p.G12C is found in 1–3% of patients with mCRC.57,61 The substitution of glycine for cysteine at position 12 results in a predominantly guanosine triphosphate-bound KRAS protein, the active form, favouring proliferation and survival of tumour cells.62,63
Recently, the isoform KRAS G12C has demonstrated to be targetable by a covalent allele-specific inhibitor. Sotorasib (AMG510) is a small molecule that specifically and irreversibly inhibits KRAS G12C in its inactive guanosine diphosphate-bound state through an interaction with one of its pockets.61 It was evaluated in a Phase I trial with 129 previously treated patients with advanced solid tumours harbouring the KRAS G12C mutation.61 In the overall trial population, of the 42 patients with CRC, only 3 (7%) presented partial response, but 28 (67%) experienced stable disease. On the other hand, 32% of the 59 patients with non-small cell lung cancer had partial response, and 56% showed stable disease. Adagrasib (MRTX849) is another KRAS G12C inhibitor under therapeutic development and it has shown promising efficacy in preclinical studies and preliminary clinical findings.64 Additional Phase I/II clinical trials are currently evaluating the efficacy of adagrasib in KRAS G12C-mutated malignancies, and an ongoing Phase III clinical trial is comparing the efficacy of adagrasib in combination with cetuximab versus chemotherapy in the second-line setting for patients with mCRC and KRAS G12C mutation (NCT04793958).65
Interestingly, patients with KRAS G12C-mutated mCRC seem to present poorer clinical outcomes compared with the patients with KRAS non-G12C mutations.66,67 A recent single-institutional study identified 187 patients with KRAS G12C from an original population of 4,685 patients with mCRC.66 When compared to a cohort of 720 patients with KRAS non-G12C mutations, these 187 patients had shorter OS, excluding patients who had undergone metastasectomy: 21.2 months versus 31.6 months (p=0.003). Another cohort of 839 patients with mCRC also found an inferior OS in G12C population compared with the non-G12C: 25.9 months versus 35.8 months (HR: 1.55; 95% CI: 1.08–2.24; p=0.018), which was confirmed by multivariate analysis (HR: 1.81; 95% CI: 1.20–2.70; p=0.04).67 Correlative findings also demonstrated that this subgroup of patients with mCRC show a distinct mutational profile, with higher rates of APC co-mutations compared with the patients without the G12C mutation, but lower rates of BRAF, ERBB4, NRAS, and TP53 co-mutations.
The reasons for the different efficacy of the KRAS G12C inhibitor according to the tissue of origin are not clear.61 Ongoing translational studies will be crucial in the understanding of the probable intrinsic resistance of KRAS G12C-mutated mCRC to the KRAS G12C inhibitors as monotherapy, and for the design of clinical trials evaluating the combination of these inhibitors with other therapeutic strategies.
HER2
Comprehensive molecular characterisation of CRC and the greater availability of next-generation sequencing in tumour genomic profiling have demonstrated that HER2 amplification is found in approximately 5–10% of patients with RAS wild-type mCRC.7,8,27 This molecular abnormality is predominantly identified in patients with RAS/BRAF wild-type patients, who may harbour HER2 amplification primarily or secondarily as a mechanism of resistance to anti-EGFR therapy.58,68-70 Exploratory analyses suggest that patients with RAS wild-type who harbour HER2 amplification derive lower, if any, benefit from anti-EGFR therapy.7,8 Based on the successful experiences of anti-HER2 therapy in HER2-amplified breast and gastric cancers, the identification of this molecular abnormality in mCRC prompted the evaluation of anti-HER2 therapy in clinical trials.
HERACLES was the first clinical trial addressing the efficacy of anti-HER2 therapy in HER2-positive mCRC.71 This proof-of-concept Phase II study was comprised of 27 treatment-refractory patients, of which 30% presented an objective response to the combination of trastuzumab plus lapatinib, and an additional 44% had stable disease. The subsequent MyPathway Phase II study showed that 32% of the 57 heavily pre-treated patients with HER2-positive mCRC had objective response to the combination of trastuzumab plus pertuzumab.72 The Phase II TAPUR basket trial evaluated the same combination of anti-HER2 therapy in 28 previously treated patients with HER2-positive mCRC, and 14% of the patients demonstrated objective response.73 Impressive findings were demonstrated by the preliminary results of the Phase II MOUNTAINEER study, which evaluated the combination of tucatinib plus trastuzumab. Of the 22 evaluable previously treated patients with HER2-positive mCRC, 55% presented objective response.74 Likewise, promising data have also been presented by DESTINY-CRC01 trial, which showed 45% of ORR with trastuzumab deruxtecan in 53 patients with previously treated HER2-positive mCRC.75 Taken together, these initial clinical trials have demonstrated that HER2 is a viable therapeutic target in mCRC, with encouraging efficacy data of the dual anti-HER2 therapy in treatment refractory patients. However, the FDA has not yet approved anti-HER2 therapies for mCRC in the USA. The results of the ongoing randomised clinical trial SWOG1613 evaluating the combination of trastuzumab plus pertuzumab in RAS/BRAF wild-type patients are eagerly awaited (NCT03365882),76 as well as the results of the Phase II DESTINY-CRC02 trial, with more data of trastuzumab deruxtecan in mCRC (NCT04744831).77
NTRK, ALK, and ROS1
NTRK are genes that encode the tropomyosin receptor kinase (Trk) family, which is comprised of three transmembrane proteins, TrkA, TrkB, and TrkC receptors, which are encoded by the NTRK1, NTRK2, and NTRK3 genes, respectively.78 The signal transduction pathways activated by these receptors are associated with proliferation, differentiation, and survival in normal and neoplastic neuronal cells.79 Gene fusions of the NTRK are the main molecular abnormalities with known oncogenic and transforming potential.80 Based on a study with 408 patients with CRC, it is estimated a prevalence rate of 0.5% of this gene fusion.81 Efficacy of larotrectinib in NTRK fusion-positive mCRC was demonstrated in a basket trial with 55 patients with solid malignancies, of which four had mCRC.82 Three patients presented tumour shrinkage and one had stable disease. Entrectinib, a pan-Trk inhibitor, has also been demonstrated to be effective in this subset of patients.83 Other gene fusions, such as those involving ALK and ROS1, are rarely found in mCRC, but, once present, they portend a poorer prognosis.84,85 Targeted therapies, including entrectinib, have been effective in this subgroup of patients with mCRC.86
CONCLUSIONS
Meaningful precision-based advancements in the therapeutic options for mCRC have been challenging and slow to realisation. Comprehensive molecular profiling and circulating tumour DNA highlights a markedly heterogeneous disease at the genomic, epigenomic, and transcriptomic levels; however, to date, they only reflect a low frequency of actionable alterations. For almost two decades, clinical applicability of precision oncology in mCRC was limited to the identification of RAS mutations as predictive biomarkers of resistance to the use of anti-EGFR monoclonal antibodies. However, novel therapeutic targets have emerged in recent years, refining the landscape of systemic therapy of the disease. The benefit of ICIs in patients with MSI-H and in those with POLE mutations or high TMB, the combination of BRAF with EGFR inhibition in patients with BRAF V600 mutations, the advent of allele-specific KRAS G12C inhibitors, and the promising findings of dual anti-HER2 therapy in HER2-positive mCRC cases have ushered in a new era of precision oncology for mCRC, providing personalised treatments and sustaining hope for patients affected by this disease.






