Abstract
Dermatofibrosarcoma protuberans (DFSP) is a rare sarcoma that most commonly occurs on the trunk of adult patients. However, DFSP can present congenitally, and this rarity may lead to misdiagnosis or late diagnosis. The authors present a case of a congenital DFSP that was not diagnosed until the patient was 17 years old. Her tumour was successfully treated with Mohs micrographic surgery. It is important to have DFSP on the differential for atrophic plaques or violaceous to red-brown nodules, even at a young age.
Key Points
1. Dermatofibrosarcoma protuberans (DFSP) can present congenitally, though their rarity may lead to misdiagnosis or late diagnosis. It is important to have DFSP on the differential for atrophic plaques or violaceous to red-brown nodules, even at a young age.2. Mohs micrographic surgery is a well-described treatment modality for DFSP that can be used for all ages.
3. It is important to evaluate DFSP for fibrosarcomatous change as these tumours are more aggressive.
INTRODUCTION
Dermatofibrosarcoma protuberans (DFSP) is a rare, locally aggressive sarcoma that most commonly occurs on the trunk of patients.1,2 The lesion typically presents as either an indurated plaque or red-brown nodules.1 DFSP occurs most often in adults during the third to fifth decades of life; however, there are reports of DFSP in childhood and even fewer reports of congenital DFSP.2-4 The authors present a case of congenital DFSP that had a delayed diagnosis and subsequent treatment with Mohs micrographic surgery (MMS).
CASE
An otherwise healthy 17-year-old female presented for evaluation of a congenital plaque on her chest. Her mother noted the plaque had been present since birth and had grown with the patient. The patient reported this previously asymptomatic lesion had recently become sore, leading her to seek evaluation. There was no family history of immunosuppression. On examination, there was a freely movable red-brown atrophic plaque 3.1×2.4 cm on the right chest (Figure 1). The remainder of her examination was unremarkable. Given the unusual congenital plaque with the new-onset of symptoms, shared decision-making was utilised to perform a punch biopsy of this lesion. The differential at this time included atrophoderma, DFSP, and medallion-like dermal dendrocyte hamartoma. The results of this biopsy demonstrated a cellular dermal and subcutaneous spindle cell proliferation (Figure 2). On staining, there was strong positivity for the CD34 antigen (Figure 3). A fluorescence in situ hybridisation (FISH) analysis was performed, which detected a platelet-derived growth factor beta (PDGFB) gene rearrangement. These constellations of findings were consistent with a diagnosis of DFSP, and MMS was elected for treatment.
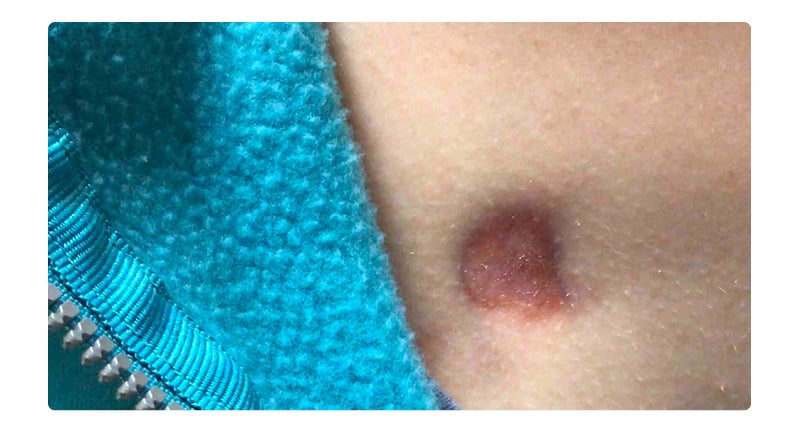
Figure 1: Red-brown atrophic plaque over the right chest.
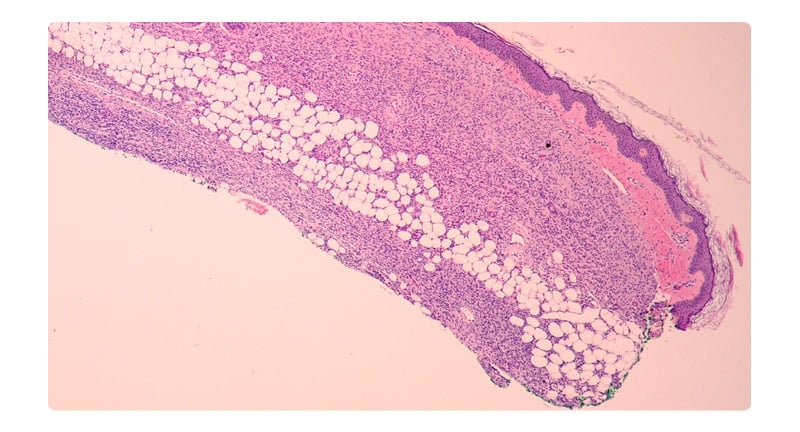
Figure 2: Haematoxylin and eosin at 4x demonstrates a dense spindle cell neoplasm infiltrating dermis and subcutis.
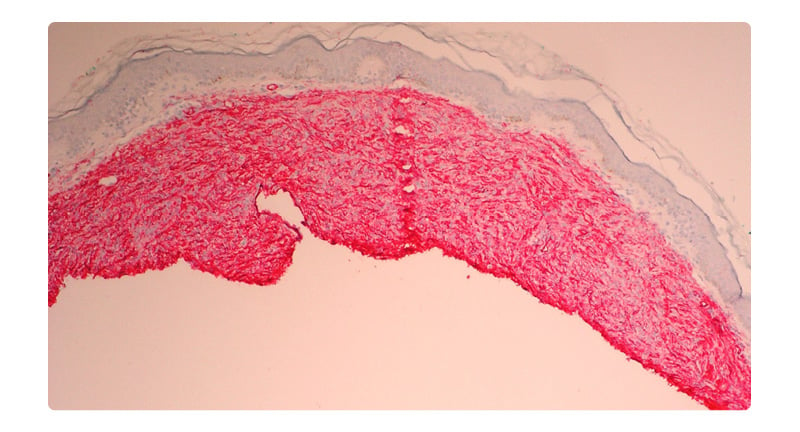
Figure 3: Immunohistochemistry for CD34 at 4x is diffusely positive in the spindle cell neoplasm.
On the day of the procedure, the patient continued to report pain from the lesion. On exam, there was a 3.1×2.4 cm atrophic plaque on the right chest with retained sutures from the punch biopsy. There was no lymphadenopathy. MMS required three stages to clear the tumour, and the site was closed with a complex primary closure. The patient has since been healing well without complications.
DISCUSSION
DFSP is a rare sarcomatous malignancy that has an overall estimated incidence of 0.8 per million persons per year.2-4 Previous reports demonstrate that children less than 16 years old account for approximately 6% of total DFSP cases, with congenital cases being even scarcer.4 The authors’ report highlights another rare case of congenital DFSP and a striking delay of diagnosis until the age of 17. A review of the literature on congenital DFSP from 2009 found that the most common presentation of congenital DFSP are nodular plaques of varying colours, but they noted that its presentation can be variable.3 This variation was demonstrated by the authors’ patient presenting with an atrophic plaque initially thought to be atrophoderma given its appearance. It is important for clinicians to recognise that congenital DFSP can occur and have differing features.3 There should be a low threshold to biopsy suspicious congenital lesions, as delay in diagnosis or misdiagnosis may lead to the continued growth of the malignancy and initiation of symptoms (as with this patient), as well as the potential for inappropriate treatments for incorrectly suspected benign congenital lesions; these impacts of delayed diagnosis may also complicate later definitive surgical treatment for a DFSP.3
On histopathology, DFSPs are composed of spindle cells with scant cytoplasm and little pleomorphism, and they are often CD34 positive.2 They are arranged in a storiform configuration and extend into the subcutaneous fat in a ‘honeycomb’ pattern.5 The aetiology of both acquired and congenital DFSP is unknown; however, many DFSPs also demonstrate a chromosomal translocation between chromosomes 17 and 22, which may be a pathogenic factor.2,3 This rearrangement leads to the fusion of PDGFB and collagen type 1A1 genes (COL1A1).2,3 This rearrangement in the authors’ patient could be evaluated by utilising fluorescence in situ hybridisation analysis.2 In the aforementioned review, they found that of 13 congenital DFSP cases tested for a PDGFB translocation, nine were positive (69%).3 The authors’ case illustrates another example of congenital DFSP having PDGFB translocation positivity and exhibits the importance of performing this analysis if there is diagnostic uncertainty in order to potentially support a diagnosis of DFSP.
Further, when evaluating DFSP histopathologically, it is important to evaluate the tumour for fibrosarcomatous (FS) change. FS under the microscope will demonstrate high-grade features including cellular atypia, increased mitoses, and a ‘herring-bone’ or fascicular growth pattern.5 It is important to evaluate for FS change of DFSP as these tumours are more aggressive with lower recurrence-free survival and increased potential for metastasis.5 The authors’ patient had no evidence of FS change on initial biopsies or final MMS procedure.
MMS is the preferred treatment modality for DFSP of all ages given lower recurrence rates, greater amounts of tissue sparing, and less overall number of surgeries.2-4 DFSP often have an unpredictable subclinical extension, and MMS provides real-time direct visualisation of the tumour over wide local excision. This is exemplified by the authors’ patient requiring three stages of MMS for clearance. In the 2009 review, they found that for congenital DFSP treated with MMS, the clearance rate was 100% (average follow-up: 4.3 years), and for the tumours treated with wide local excision, the clearance rate was 89% (average follow-up: 1.9 years).3 They also found that excised margins were smaller with MMS over wide local excision.3 The authors’ adolescent patient was able to have tumour clearance with the greatest amount of tissue sparing in a sensitive site, demonstrating the benefit of MMS in these cases.
CONCLUSION
DFSP can present congenitally, though their rarity may lead to misdiagnosis or late diagnosis. It is important to have DFSP on the differential for atrophic plaques or violaceous to red-brown nodules, even at a young age. Biopsy and subsequent histopathological testing are key in the diagnosis of DFSP. MMS is the preferred treatment of choice for congenital DFSP given higher clearance rates and smaller excised margins.






