Abstract
Epithelioid glioblastoma is a rare and aggressive variant of glioblastoma that is common in children and young adults. This variant frequently has a BRAF V600E mutation, and in recent years, this is often treated with BRAF and mitogen-activated protein kinase kinase inhibitors. An 18-year-old female initially presented with headaches and vomiting. They were diagnosed with an epithelioid glioblastoma and treated with temozolomide chemoradiotherapy. Upon progression, they had to redo surgery and then received dabrafenib and trametinib. They had one last surgery shortly before fatal tumour progression. Retrospective molecular analysis of three tumour specimens showed a PTEN mutation that arose upon first progression, but was not there initially. There were no new tumour mutations after initiation of dabrafenib and trametinib. The acquired PTEN mutation may have conferred resistance to dabrafenib and trametinib. This case highlights the potential importance of early treatment with BRAF and mitogen-activated protein kinase kinase inhibitors in high-grade BRAF V600E-mutated gliomas, ideally before the tumour develops resistance to targeted therapy.
Key Points
1. The combination of BRAF and mitogen-activated protein kinase kinase inhibitors has recently shown to help treat some patients with BRAF-mutated high-grade glioma, but it is not entirely clear why some patients respond, while others do not.2. The authors report a case of a young female with a BRAF V600E-mutated epithelioid glioblastoma treated with temozolomide chemoradiotherapy. Upon tumour recurrence a PTEN loss of function mutation became apparent and they did not respond to dabrafenib and trametinib.
3. Loss of function PTEN mutations may confer resistance to dabrafenib and trametinib in glioma, so earlier targeted therapy treatment (before PTEN mutation occurs) might improve outcomes.
INTRODUCTION
Proto-oncogene B-Raf or BRAF is a gene found on chromosome 7q34 in humans that encodes the protein B-RAF, a member of the RAF group of serine/threonine protein kinases.1 Once activated, BRAF goes on to phosphorylate the downstream mitogen-activated protein kinase kinase (MEK) protein as part of the mitogen-activated protein kinase signalling pathway.² This pathway helps to regulate growth, proliferation, differentiation, and apoptosis of cells.2
Activating mutations of the BRAF gene lead to uncontrolled cell growth. This can be found in non-malignant lesions such as low-grade gliomas and nevi.3,4 Usually second hits are required to circumvent oncogene-induced senescence and induce the formation of malignant tumours.
Mutations are thus common oncogenic triggers as it has been estimated that BRAF gene mutations are involved in 8% of all malignancies, the most frequent of which are melanoma and thyroid cancers.5 Activating mutations of the BRAF gene include fusions and point mutations. The former drives oncogenic changes through genomic rearrangement. Specifically, the 3’ part of the BRAF gene that encodes for the kinase domain is placed at the 5’ position after another gene.6 The auto-inhibitory area of the BRAF gene is consequently lost, triggering constitutive activation of the oncoprotein.6 The most common point mutation is a substitution of thymine for adenine at nucleotide position 1799 within the BRAF gene, which results in the substitution of valine for glutamic acid.7 This also causes constitutive activation through the failure of inhibition of BRAF activity. These BRAF alterations are found in glioblastoma (GBM), pleomorphic xanthoastrocytoma, and low-grade gliomas in children.8 Moreover, it was also found that clinically distinct subsets of secondary high-grade glioma are defined by the presence of BRAF V600E mutation.9 Lastly, the BRAF V600E point mutation is common in epithelioid GBM (E-GBM).10
BRAF V600E commonly occurs concurrently with the silencing of the PTEN tumour suppressor gene.11 For example, more than 30% of all melanomas also have a PTEN loss of function mutation.12 The PTEN gene encodes for a protein that has scaffolding functions in a cell and dephosphorylates lipids such as phosphatidylinositol-3,4,5-trisphosphate.13 By acting as the phosphatase for this lipid, the protein antagonises the phosphoinositide 3-kinase (PI3K)/Akt pathway, resulting in inhibition of cell proliferation, therefore regulating the cell cycle.13 The loss of PTEN results in aberrant activation of the PI3K/Akt pathway. This is an oncogenic trigger, as the PI3K/Akt signal transduction pathway when functioning normally is responsible for controlled cell proliferation, growth, and survival.14
PTEN loss of function mutation is also commonly found in GBM, as hemizygous or homozygous deletion have been found to occur in more than 90% of patients with GBM.15 The impact of PTEN loss of function mutations in survival of patients GBM is not definitively known as some studies state that the mutation is closely associated with the prognosis,16 while others state that there is no significant association.17
The combination of both loss of PTEN and BRAF V600E point mutation has been reported in cases of GBM, giant cell GBM, rhabdoid GBM, and supposed Grade 3 oligodendroglioma.15 Of interest are the range of alterations that can occur in combination with BRAF V600E as cancers transform. Alterations that can occur in combination with BRAF V600E may point to why the acquisition of this alteration may be a mechanism of resistance.
Below is a novel case of a patient with BRAF V600E-mutated E-GBM who later developed PTEN loss of function mutation only after initial temozolomide chemoradiotherapy. This is the first case that suggests PTEN loss may mediate resistance to dabrafenib and trametinib in a BRAF V600E-mutated high-grade glioma.
CASE PRESENTATION
A previously healthy 18-year-old female developed headaches, sometimes followed by vomiting. Family history was non-contributory and the patient was not on medications. These symptoms worsened over 6 months. The patient then developed daily vomiting for 1 week and was admitted to hospital. They underwent CT imaging, which demonstrated an expansile heterogeneously enhancing right temporal lesion. Their MRI imaging revealed areas of susceptibility artifact and possible calcification. There was a mass effect that compressed the surrounding sulci, the right lateral ventral, and third ventricle. There was 10 mm of midline shift.
The patient was taken to the operating room for an initial craniotomy for tumour resection. There was a gross total resection of the tumour (Figure 1). On a pathologic examination the patient was diagnosed with an ‘anaplastic ganglioglioma’. They were treated with focal radiotherapy (60 Gy in 30 fractions) with concomitant temozolomide. The patient experienced persistent neutropenia after concomitant temozolomide and consequently did not receive adjuvant temozolomide.
Follow-up MRI 10 months later revealed tumour progression (Figure 1). The patient underwent a second resection approximately 1 month following initial resection. Pathologic examination at this time revealed E-GBM and a BRAF V600E mutation was identified.
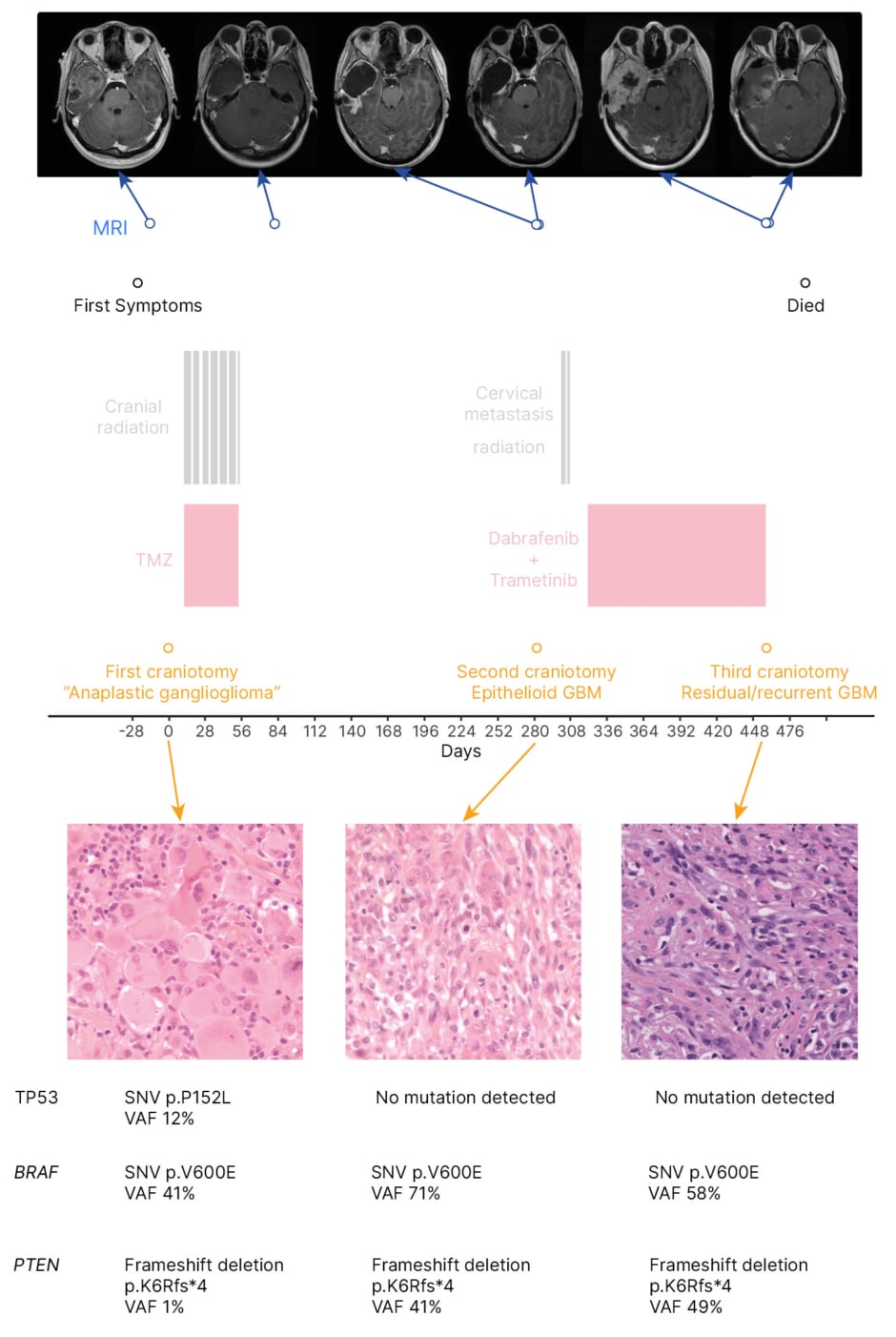
Figure 1: Timeline of clinical, radiological, and pathological information for an 18-year-old patient with epithelioid glioblastoma treated with temozolomide chemoradiotherapy, followed by dabrafenib and trametinib.
GBM: glioblastoma; SNV: single nucleotide variant; TMZ: temozolomide; TP53: tumour protein 53; VAF: variant allele frequency.
In retrospect, the patient was found to have a PTEN loss of function mutation that was not present at initial surgery, but was present at their second surgery, before receiving dabrafenib and trametinib. This loss of function was the result of a frameshift deletion. The patient’s CDKN2A status was unknown. The first tumour sample was also shown to have a single nucleotide variant in TP53 p.P152L, which was not detected in later samples. This variant when present in the germline does not tend to result in the development of brain tumours. It has been seen on rare occasions in gliomas.18 In this patient, the variant may have been incidental and not pathogenic.
The patient developed a drop metastasis to the cervical spine and consequently received focal radiotherapy (20 Gy in five fractions) to the cervical spine 20 days following the second resection. They were approved for compassionate access to dabrafenib and trametinib, and began taking these 1 month following the second surgery. There was no response to the treatment and the primary tumour developed clinical progression 4 months after receiving compassionate dabrafenib and trametinib. The treatments were stopped approximately 6 months after the second resection due to further disease progression. The patient developed uncal herniation as a result of recurrent GBM, which required a third resection approximately 5 months after their second resection (Figure 1). The patient died 1 month after the third resection, 17 months after initial diagnosis.
DISCUSSION
E-GBM is a variant of GBM that is more common in adolescents and young adults.19 The cancer is differentiated from other GBM variants by frequent dissemination to the leptomeninges.20,21 Genetic analysis studies reveal that close to 50% of E-GBM have a BRAF V600E mutation.22 As such, BRAF inhibitors such as dabrafenib and MEK inhibitors such as trametinib have induced clinical responses.23 For example, in one case of E-GBM treated with this combination therapy, the patient achieved clinical and radiological stability for 53 months after starting treatment.24,25
This case represents the first description of PTEN mutation in a patient with BRAF V600E-altered E-GBM. There are however two cases documented in The Cancer Genomic Atlas (TCGA) with similar tumour molecular alterations (i.e., GBM with BRAF V600E and PTEN silencing mutations).24,25 The first patient had a left parietal GBM and a PTEN frameshift deletion. They were treated with the Stupp protocol.24,25 Upon first progression, they received irinotecan and bevacizumab. After the second progression, they received temozolomide and re-irradiation. The patient died 12 months after diagnosis.24,25 The second TCGA patient also had a left parietal GBM, a PTEN frameshift deletion, and underwent Stupp protocol. The patient developed tumour progression but details about latter line therapies and survival time are unavailable.24,25 Notably, the TCGA listed four patients with GBM who had the BRAF V600E mutation, and they had survival times between 3–46 months. The average survival time for patients with BRAF V600E mutations without a concurrent PTEN loss of function mutation was 17.25 months, and the median was 10.00 months.24,25 Comparatively, the patient described in this case report had a survival time of 12 months from the time of first diagnosis. Interestingly, a search of the TCGA database for low-grade gliomas with concomitant BRAF and PTEN mutations revealed no hits. These double hits seem to mark high-grade behaviour and histology.
As the combination of these mutations has not been widely studied in E-GBM, much of what is known stems from investigations of the effects of PTEN and BRAF V600E in cancers outside of the central nervous system. The prolonged activation of the mitogen-activated protein kinase pathway secondary to the BRAF V600E mutation and the activation of the PI3K pathway secondary to the PTEN mutation results in melanoma progression and metastasis development.26 This combination has been previously demonstrated to result in 100% penetrance of melanomas that metastasise to the lungs and lymph nodes.26 Moreover, PTEN loss of function is associated with intrinsic resistance to BRAF and MEK inhibitor drugs, therefore limiting the use of these medications in patients with melanomas with both BRAF V600E and PTEN mutations.11 The authors hypothesise that this is what happened to the patient in the present case with PTEN loss leading to intrinsic resistance to dabrafenib and trametinib. Cancers in other parts of the human body have also been shown to involve the combinatory effect of PTEN and BRAF mutations. For example, these mutations were found to drive the expression of c-Myc, confer resistance to castration, and drive metastasis to lymph nodes as well as the lungs in mice models of advanced prostate cancer.27 Additionally, these mutations were reported to confer acquired resistance to BRAF inhibitors dabrafenib/trametinib in a patient with squamous cell lung cancer.28
What makes this reported case notable is the implications for management of patients with E-GBM. First, the patient may have achieved clinical and radiological stability had they been placed on dabrafenib and trametinib after initial surgery. Additionally, the patient may have benefited from being placed on a PI3K inhibitor, as the PI3K/Akt pathway is aberrantly activated by a PTEN loss of function mutation.
Similar to this, further investigations are required to determine if combination therapy co-targeting the BRAF and PI3K/Akt pathway is of benefit for patients with E-GBM with co-occurring BRAF and PTEN mutations. This form of therapy has already shown to be effective in treating melanomas that metastasise to the brain in both in vivo and in vitro experiments.29 In one study, the researchers targeted melanoma brain metastasis cell lines derived from patients with PI3K inhibitor buparlisib, as well as MEK inhibitor trametinib, and compared the effect of combination treatment with monotherapy. The investigators found that relative to monotherapy of trametinib, combination therapy increased apoptosis and diminished tumour growth in vivo.29 However, a potential obstacle in using this strategy in patients with E-GBM is that there are very few PI3K inhibitor drugs that cross the blood–brain barrier and are not neurotoxic.30 If this obstacle can be overcome, co-targeting both pathways may hold promise for new treatment for future patients with E-GBM.
Further studies are required on the temporal evolution of molecular alterations in BRAF V600E-altered high-grade gliomas. This tumour evolution provides a basis for a trial to include targeted agents, such as BRAF and/or MEK inhibitors, in the initial management of BRAF V600E-mutated high-grade gliomas at diagnosis, rather than solely at progression (as with this patient).
As noted above, BRAF and PTEN mutations occur in combination in many different types of tumours and seem to be more aggressive than BRAF V600E alone. Future investigations will be required to explore tumour evolution in this context and understand mechanisms of drug resistance. This may have broader implications for other BRAF-mutant brain tumours and is significant to understand for high-grade glioma to design rationale clinical trials moving forwards. It is also crucial to understand low-grade glioma as there is risk of transformation to high-grade glioma, and understanding relevant biomarkers may allow earlier and more aggressive intervention.
Lastly, it will be interesting to discover whether the findings of the present case apply to other BRAF V600E-mutated high-grade glioma, or even low-grade glioma.
CONCLUSION
In conclusion, this case report highlights the novel finding of an acquired PTEN loss of function mutation in a patient with a BRAF V600E-mutated E-GBM. The combinatorial effect of these mutations likely resulted in the development of intrinsic resistance to BRAF and MEK inhibitor medications, dabrafenib and trametinib, respectively. Current understanding of these mutations in combination stems from the presence in other malignancies such as melanoma, papillary thyroid cancer, and squamous cell lung cancer. Further investigations are required to fully delineate the implications of these mutations on the management of patients’ BRAF V600E glioma.