
Meeting Summary
Laura Campbell from Orchard Therapeutics, London, UK, opened the session by explaining the objectives of the symposium and providing some background information on metachromatic leukodystrophy (MLD). MLD is a rare and life-threatening inherited disease of the body’s metabolic system. MLD is caused by a mutation in the ARSA gene, which results in the accumulation of fats called sulfatides in the brain and other areas of the body, including the liver, gallbladder, kidneys, and spleen. Over time, the nervous system is damaged and children with MLD experience progressive neurological symptoms, including motor, behavioural and cognitive regression, severe spasticity, and seizures. Patients with MLD gradually lose the ability to move, talk, swallow, eat, and see. MLD is estimated to occur in approximately one in every 100,000 live births.1,2 The prognosis for MLD is extremely poor. Most children within the late infantile (LI) form die by the age of 5 years; the juvenile (JU) form progresses to death within 10–20 years; and those affected by the adult form typically die 25 years following the onset of symptoms.1 Approval of Libmeldy (atidarsagene autotemcel, [arsa-cel]; Orchard Therapeutics, London, UK), a gene therapy containing an autologous CD34+ cell enriched population, which contains haematopoietic stem and progenitor cells transduced ex vivo using a lentiviral vector encoding the human ARSA gene, for the treatment of early-onset MLD,3 opens up tremendous new possibilities for eligible children with MLD faced with this devastating disease, where previously no approved treatment options existed. Libmeldy is the first product approval for Orchard Therapeutics, a global gene therapy leader dedicated to rare diseases through the development of gene therapies.4 Francesca Fumagalli from the Paediatric Immunohematology Unit and Department of Neurology, Ospedale San Raffaele – Telethon Institute for Gene Therapy (OSR-TIGET), Milan, Italy, shared the evidence on the efficacy and safety of Libmeldy in patients with MLD. The clinical trial investigating Libmeldy started more than 10 years ago at OSR-TIGET. Campbell closed the symposium by providing several educational resources to support clinicians managing children with MLD.
“MLD is a heart-breaking disease that causes immeasurable suffering and robs children of the chance of life. As a community, we have been desperate for a treatment for young patients with MLD, and we are incredibly excited to now have such a ground-breaking option approved in the European Union (EU).” Georgina Morton, Chairperson of ArchAngel MLD Trust4
Libmeldy is indicated for the treatment of MLD characterised by biallelic mutations in the ARSA gene, leading to a reduction of the arylsulfatase A (ARSA) enzymatic activity in children with LI or early juvenile (EJ) forms, without clinical manifestations of the disease, and in children with the EJ form, with early clinical manifestations of the disease, who still have the ability to walk independently and before the onset of cognitive decline.
Early Signs and Symptoms of Metachromatic Leukodystrophy
Laura Campbell
MLD is caused by an accumulation of sulfatides in various organs and predominantly the central nervous system (CNS). Patients have an initial period of normal development, then experience developmental delay or stagnation (Figure 1).5 When ARSA is deficient, sulfatides build up to high levels in the protective myelin sheath surrounding nerves, disrupting the myelin structure, and causing demyelination to occur in both the CNS and in the peripheral nervous system (PNS). The sulfatides will also build up in the visceral organs (such as the kidneys and gallbladder).6,7 At this point, patients deviate from the previous development path and the normal trajectory of their peers, into a plateau phase. For example, they may stop progressing in their efforts towards walking; a patient with LI MLD may never learn to walk. After this period, patients begin to lose previously learned skills and will begin to exhibit more obvious symptoms of disease such as muscle weakness and neurocognitive changes. After these early signs and symptoms, patients then enter a rapidly progressive phase where they have rapid developmental regression and disease progression within months. After a noticeably short period of time, months or, in some cases, a few years, these patients will then enter a phase of severe neurological disability.5
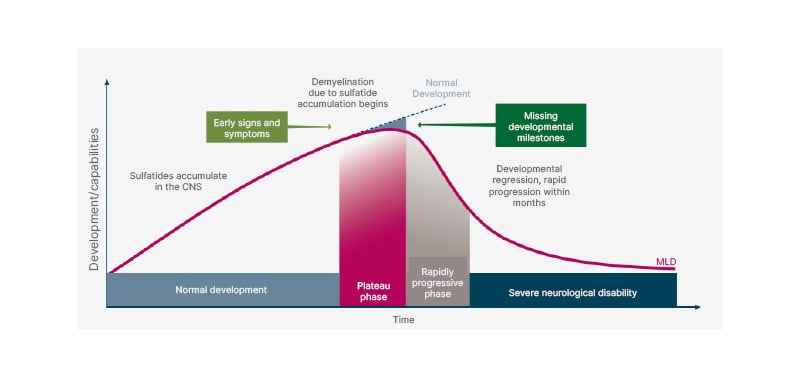
Figure 1: Clinical course of metachromatic leukodystrophy.
Adapted from von Figura K et al.5
CNS: central nervous system; MLD: metachromatic leukodystrophy.
The most common early signs and symptoms reported by caregivers in patients with LI MLD include motor symptoms such as difficulty in walking/running, clonus/tremor, co-ordination/balance, and unilateral muscle weakness. Initial symptoms reported in patients with JU MLD include a mix of motor and developmental/behavioural problems such as difficulty in walking/running, change in behaviour/personality, and lack of awareness.8 These initial symptoms often manifest as a lack of developmental progress, or what is termed ‘persistent toddler’ or ‘developmental stagnation’, where a child had previously reached developmental milestones normally, but is now no longer progressing as expected, which should trigger immediate further investigation. The indication from parents and other caregivers that something is amiss is an important tool in recognising the early stages of MLD. The reason that this is so important is that once MLD has taken hold in a patient, demyelination and progression of disease is extremely rapid and can lead to early disability, as well as early death.9 Therefore, it is vital that clinicians are aware of the early signs and symptoms of MLD in order to be able to identify these patients early and refer them to a specialist for appropriate treatment to improve morbidity and mortality outcomes for patients.
“[Patients with LI form of MLD] rapidly experienced psychomotor deterioration, bulbar dysfunction, and seizures, with loss of trunk control and dysphagia occurring concomitantly, and resulting in a substantially reduced survival.”9
The Trial Results of Libmeldy (Atidarsagene Autotemcel) for the Treatment of Early-Onset Metachromatic Leukodystrophy
Francesca Fumagalli
MLD is a rare, autosomal-recessive lysosomal sphingolipid storage disorder disease, caused by a mutation in the ARSA gene.1,10-12 This leads to the accumulation of toxic substrates, sulfatides, in different tissues, but in particular in myelin-forming cells of the CNS and PNS, resulting in progressive demyelination and neurodegeneration.9 The disease can be considered a phenotypic continuum caused by common underlying pathophysiological mechanisms, but it has historically been classified in different variants according to the severity, and based on a combination of the age of onset of symptoms and genotype. Three main clinical phenotypes have been described: the LI (age at symptom onset: 0–30 months) form (50% of cases), usually associated with two 0 alleles, and characterised by early motor impairment, rapid psychomotor regression, and premature death (mean age of death: 4.2 years). JU (age at symptom onset: 30 months–17 years) form (35% of cases) is characterised by a combination of motor and cognitive/behavioural deficits at onset and a more heterogenous progression than LI and premature death (mean age of death: 17.4 years). Patients with JU MLD are commonly subdivided into EJ and late juvenile (LJ), and together LI and EJ MLD are termed ‘early-onset MLD’ (onset prior to seventh birthday). The disease course of early onset MLD is very rapid, with a fatal outcome within a few years after symptom onset. Adult onset MLD (age at symptom onset: >17 years) form (15% of cases) presents with cognitive decline, behavioural and psychiatric disturbances, and with variable outcomes (mean age of death: 43.1 years).1,11-15
The Gross Motor Function Classification in MLD score (GMFC-MLD) is commonly used to describe the progressive loss of motor function in children with MLD,2 and Figure 2 clearly describes how rapidly untreated children lose their ability to walk independently (corresponding to Level 2) and reach a very severe disability with the loss of trunk control (Level 5) a few years after symptom onset. In parallel with motor decline, severe spasticity, epilepsy, dysphagia, as well as loss of speech, hearing, and sight can occur.16 For that reason, the treatment of these children is usually only supportive, and based on palliation of symptoms in the symptomatic phase. The most important challenge in the development of effective treatments for MLD is the need to cross the blood–brain barrier and target the CNS.10,14,17,18 Treatment options for MLD have, therefore, been limited. Allogeneic haematopoietic stem cell transplantation (HSCT) has been used in other diseases for decades and is based on the principle of the migration of physiologically functional donor cells to the CNS, which provide metabolic cross-correction. However, reports of allogeneic transplant outcomes for MLD in the medical literature are relatively few and conclusions are mixed. HSCT has shown some efficacy in modifying disease course in later onset (late juvenile and adult) variants, but has not demonstrated benefits in early onset MLD (LI and EJ), which are the more aggressive and most common forms, even when administered pre-symptomatically.14,17 In the past few years, advances in translational research have brought hope to patients affected by MLD, with different therapeutic strategies showing promising results in slowing disease progression.19-25 However, the evaluation of efficacy of such treatments is sometimes hindered by the complexity and variability of the disorder.2,26,27
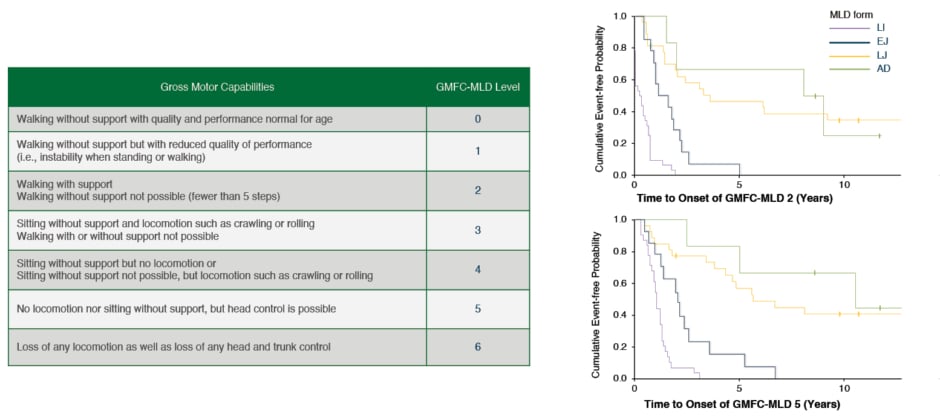
Figure 2: GMFC-MLD level description and motor disease progression in MLD
Adapted from Kehrer C et al.2,14
AD: adult; EJ: early juvenile; GMFC-MLD: Gross Motor Function Classification for metachromatic leukodystrophy; LI: late infantile; LJ: late juvenile; MLD: metachromatic leukodystrophy.
Libmeldy is an ex vivo autologous haematopoietic stem cell gene therapy. The first in human clinical trials with arsa-cel started in 2010, and Libmeldy was approved for use by the European Commission in children with LI or EJ forms of MLD on 17th December 2020.3 This was closely followed by approval from the Medicines and Healthcare products Regulatory Agency (MHRA) on 1st January 2021.28 Libmeldy is currently the only approved treatment for MLD. With Libmeldy, a patient’s own HSCs are collected, and functional copies of the ARSA gene are inserted into the genome of the HSCs using a self-inactivating lentiviral vector. Then, these genetically modified cells are infused back into the patient. Gene-corrected HSCs are able to migrate across the blood–brain barrier into the brain, engraft, and express functional enzyme, which has the potential to persistently correct the underlying disease with a single treatment.3
The approval of Libmeldy was based on the results of the NCT0156018229 and NCT0339298730 clinical trials and patients treated in expanded access frameworks (EAF). As of June 2021, 39 patients have been treated in the clinical development program with Libmeldy (20 in NCT01560182; 9 in EAFs; and 10 in NCT03392987). Eligible patients included those with early-onset MLD, with either pre-symptomatic LI variant or pre- or early-symptomatic EJ variant. In NCT01560182, early-symptomatic-EJ was defined as having an intelligence quotient (IQ) of ≥70 and the ability to walk independently for ≥10 steps.29 Based on preliminary results, this definition was amended to stricter criteria to ‘free from cognitive impairment’ (IQ: ≥85) and able to walk independently without support (GMFC-MLD Level 0 or 1) in NCT03392987.30 The objectives of these clinical trials were to evaluate the safety and efficacy of Libmeldy. The primary efficacy endpoints of NCT01560182 were improvement in Gross Motor Function Measure (GMFM) score, as well as a restoration of ARSA activity at 2 years after treatment compared with baseline. The studies also evaluated the short- and long-term safety of gene therapy, treatment effects on cognitive function, and brain atrophy and demyelination in the CNS and PNS (assessed by MRI and nerve conduction velocity).
There were 16 patients with the LI form (15 pre-symptomatic, and one who became symptomatic between enrolment and treatment) and 13 patients with EJ MLD (five pre-symptomatic and eight early-symptomatic) participants in NCT01560182 and the EAFs with a median length of follow-up of 3.16 (0.64–7.51) years. Additional data, particularly on safety and pharmacodynamics, are available for patients more recently treated with the cryopreserved formulation. The results in the 29 participants treated with the fresh formulation were compared with an untreated natural history cohort of 31 patients with early-onset MLD, who were followed with the same clinical and instrumental tools. All 29 patients showed persistent and stable engraftment of corrected cells. At 1-year post-treatment, the mean percentage of lentiviral vector-positive bone marrow progenitor cells was 55% (range: 20–100%). Stable vector copy number in CD34+ cells was seen throughout the follow-up period. Notably, the transduced progenitors were detectable starting from 1 month post-gene therapy. This resulted in a rapid increase of ARSA activity in peripheral blood that stabilised within 3–6 months post-gene therapy at normal to supranormal levels in all patients. The study confirmed that ARSA activity in the cerebrospinal fluid, undetectable at baseline, was detected from the first measurement performed at 3 months after gene therapy treatment, reached normal levels by 6–12 months post-gene therapy, and remained within the normal reference range up to the longest follow-up available (60 months).31
The effects of Libmeldy treatment on motor function was assessed using the GMFM and GMFC-MLD. The GMFM is a motor function scale that ranges from 0 to 100, where 100 is normal function in a child of 5-years-old. A co-primary efficacy endpoint was an improvement in >10% in GMFM at 2 years post-treatment, compared with the age and disease subtype-matched natural history control (Figure 3).31 A statistically significant difference far exceeding the pre-defined 10% threshold was found in both patients with the LI and EJ MLD at 2 years (p<0.001 and p=0.036, respectively), which further increased at 3 years (p<0.001 for both LI and EJ), compared with the natural history cohort. Most of these patients displayed motor development similar to or slightly below normally developing children, or had stabilisation of motor function or delayed motor decline compared with the natural history group. Severe motor impairment was defined as reaching GMFC-MLD Level 5 (loss of trunk control). The study demonstrated that severe motor impairment or death was prevented or delayed compared with natural history in the majority of patients, including patients with EJ MLD who were treated in an early symptomatic phase.31
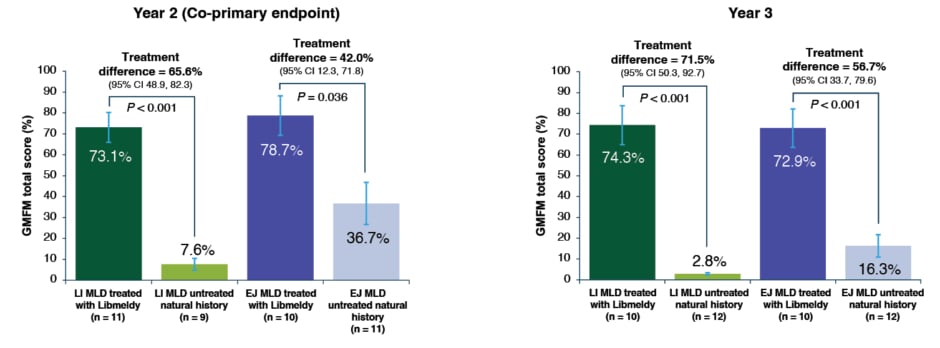
Figure 3: Gross Motor Function Measure scores in patients treated with Libmeldy versus untreated natural history
Note: vertical error bars are SE of the adjusted mean and p-values are from a two-sided 5% hypothesis test with null hypothesis of ≤10% difference.
Adapted from Fumagalli et al.31
CI: confidence interval; EJ: early juvenile; GMFM: Gross Motor Function Measure; LI: late infantile; MLD: metachromatic leukodystrophy; SE: standard error.
Most patients maintained normal cognitive functioning in terms of performance score and acquired cognitive skills in line with the chronological age. Furthermore, four out of five children with EJ MLD treated in the early-symptomatic phase, who would have fit the current approved indication at the time of treatment (able to walk independently [GMFC: 0 or 1] and before the onset of cognitive decline [IQ: ≥85]), achieved development and acquisition of cognitive abilities in the normal range during long-term follow up. The observed clinical efficacy was further supported by imaging tests using an adapted Loes score,32 to quantify the extent of white matter abnormality and the progression of brain atrophy. Brain MRI total scores for patients with LI MLD who were treated with Libmeldy stabilised at levels significantly below those observed in the natural history cohort, hence demonstrating lower levels of brain atrophy and demyelination in treated patients. Brain MRI scores also stabilised in the majority of patients with EJ MLD at lower levels than natural history patients with untreated EJ MLD, although at slightly higher levels than in patients with treated LI MLD.31
Regarding PNS involvement in MLD, the LI variant is associated with a very severe peripheral neuropathy, presenting early in the clinical course with a rapid decrease to very low nerve conduction velocities. PNS involvement in LI MLD has been refractory to correction with allogeneic HSCT.20 In contrast, in patients with LI MLD who were treated with Libmeldy, after an initial decline in the first months after gene therapy, nerve conduction velocity stabilised at statistically significant higher levels than in the untreated natural history patients in the deep peroneal and ulnar nerves (both p<0.0001) throughout the follow-up period. This suggests a positive treatment effect of Libmeldy on progressive peripheral demyelination in LI MLD.33
A detailed analysis of treatment outcomes was conducted to identify clinical factors that could influence the level of treatment benefit with Libmeldy and optimise the recommended use of the treatment. This analysis identified four treatment failures. As anticipated, in the LI group, one child who was symptomatic at treatment progressed at the same rate as the natural history group after treatment. Therefore, only patients with LI MLD without clinical manifestations of the disease are within the indicated population for treatment with arsa-cel. In the patients with early-symptomatic-EJ MLD, three patients treated with Libmeldy showed deterioration in both motor and cognitive functions comparable with that observed in untreated natural history patients, and progression of the disease led to death in two of these patients. Their clinical history showed that they either had an IQ of <85, a GMFC of ≥2 at baseline, or experienced deterioration of disease between screening and treatment, suggesting they had entered the rapid phase of disease progression before gene-modified cells were able to engraft. Taking these results into consideration, treatment with Libmeldy in patients with the EJ form of the disease is restricted to those who still have the ability to walk independently, and before the onset of cognitive decline.32 In the summary of the product characteristics of Libmeldy, it is noted that the physician will reassess for disease progression between initial evaluation and drug infusion; if the disease has worsened, the child many not benefit from Libmeldy and the doctor may recommend against treatment.32
As of data cut for this analysis, the safety of Libmeldy was evaluated in 35 patients with MLD. The median duration of follow-up in the integrated safety data set of 29 patients treated with the fresh (investigational) formulation was 4.51 (range: 0.64–7.51) years, and in the six patients treated with the cryopreserved formulation was 0.87 (range: 0.00 to 1.47) years. There were three deaths among the treated group: two related to disease progression and one due to an ischaemic stroke; none were considered to be related to Libmeldy treatment. Treatment with Libmeldy was generally well-tolerated, with most adverse events attributed to busulfan conditioning or MLD disease progression. Treatment-related adverse events included anti-ARSA antibodies, which were reported in five patients. Titres were generally low and resolved spontaneously or after treatment with rituximab, with no apparent impacts observed on clinical efficacy or safety outcomes. No malignancies occurred; however, there is a theoretical risk of leukaemia or lymphoma after treatment with Libmeldy and patients should be monitored for 15 years after treatment, in line with other gene therapies.31,32
Treatment with Libmeldy is preceded by medical interventions, namely HSC collection through bone marrow harvest or peripheral blood mobilisation with granulocyte colony-stimulating factor, with or without plerixafor followed by apheresis, and myeloablative conditioning (busulfan is recommended), which are associated with well characterised but potentially serious risks. The safety profile and product information of the medicinal products used for peripheral blood mobilisation and myeloablative conditioning should be considered, in addition to the risks linked to the gene therapy. For further (or detailed) safety information, refer to the EMA summary of product characteristics for Libmeldy.32
Libmeldy must be administered in a qualified treatment centre with experience in HSCT. The long-term efficacy and safety of Libmeldy are currently unknown. Patients are expected to enrol and be followed in a long-term follow-up study in order to better understand the long-term safety and efficacy of Libmeldy for up to 15 years following treatment. Five treatment centres are qualified in Europe: Ospedale San Raffaele, Milan, Italy; Children’s Hospital, University of Tübingen, Germany; Royal Manchester Children’s Hospital, UK; APHP Robert Debré Hospital, Paris, France; and University Medical Center, Utrecht, The Netherlands.
Fumagalli concluded that the results from this interim analysis of 35 patients demonstrated a favourable benefit-risk profile of Libmeldy in the treatment of early-onset MLD, followed for up to 7.5 years post-treatment. There have been no serious adverse events related to Libmeldy reported to date. All patients treated with the fresh formulation achieved stable levels of haematological engraftment and reconstitution of ARSA activity in peripheral blood and in cerebrospinal fluid to normal to supranormal levels. Similar results have been seen for the cryopreserved formulation and within the range of results observed for the fresh formulation. Treatment effects observed in gross motor function and cognition suggest that Libmeldy provides clinically meaningful benefit in pre-symptomatic patients with LI and EJ MLD and patients with EJ MLD in the early symptomatic stages of MLD. Libmeldy may support the prevention, stabilisation, or delay of hallmark progressive CNS damage of early-onset MLD, consistent with treatment effects observed on motor function and cognition. The long-term follow-up of patients treated with Libmeldy is ongoing.
Conclusion
There is an urgency to diagnose, refer, and treat patients with early-onset MLD as there is a limited treatment window for this fatal disease. Lack of developmental progress, when previously reaching milestones normally (i.e., the persistent toddler should be recognised early, requiring immediate further investigation and referral for appropriate life-saving treatment). There is now a beneficial treatment option available in Europe for early-onset MLD, a population for whom options were previously limited or absent. Libmeldy has a positive effect on the neurological symptoms and motor skills following a single administration. These data presented by Fumagalli clearly highlight that early treatment can bring hope to patients.
Great progress is being made in MLD research to better understand the disease, its presentation, the importance of new-born screening, and available treatments. Recently, the academic led MLD initiative used a modified Delphi procedure to achieve consensus and harmonisation on core data to be collected to support research and to facilitate compliance with regulatory requirements.34 There are several resources available to support clinicians, including the reference list at the end of this article and a series of educational webinars with world leading experts in the field, offered by Excellence in Pediatrics (EiP), which covers the diagnosis and management of MLD.35






