Meeting Summary
A masterclass initiated, organised, and funded by UCB, sought to advance understanding and expertise of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD).
MOGAD is a rare central nervous system (CNS) inflammatory disorder. It has only relatively recently been characterised as a separate entity to similar demyelinating conditions, such as multiple sclerosis (MS) and neuromyelitis optica spectrum disorder (NMOSD). Differentiating MOGAD from MS and NMOSD can be challenging, but is essential to ensure a correct diagnosis in order to guide effective treatment and management.
An increase in the availability of cell-based assays (CBA) for detection of autoantibodies directed against myelin oligodendrocyte glycoprotein (MOG) over the last decade has provided healthcare professionals with an important new diagnostic tool. However, the approach has limitations in terms of sensitivity and specificity, meaning results must be considered alongside clinical characteristics and neuroimaging. A proposed diagnostic pathway by the international MOGAD panel, published in March 2023, sets out the core clinical demyelinating events that could suggest MOGAD, when MOG-IgG testing may be appropriate, and when supporting clinical or MRI features are required to confirm a diagnosis. These consensus criteria are now being evaluated by centres around the world.
This article will summarise the talks given by key opinion leaders from across Europe and the USA during the educational event. They discussed the pathology and presentation of MOGAD, how to integrate imaging into diagnostic pathways, and current management approaches. They also looked at possible future directions, in terms of novel treatment approaches.
Introduction
MOGAD is a rare CNS disease, which has only relatively recently been characterised as a separate entity to similar demyelinating conditions, such as MS and NMOSD.1 Despite having overlapping characteristics, each of these diseases requires different treatment approaches,1,2 and making the correct diagnosis is essential to quality patient care, the speakers said. The advancing expertise in the understanding of MOGAD masterclass looked at the latest research and trends related to the understanding of the condition, as well as advances in diagnostics and management.
Speakers started the day with an overview of MOGAD, a rare CNS inflammatory disorder with a global prevalence of 0.50–3.42 cases per 100,000 people.1 They explained that demyelination associated with MOG antibodies can result in variable clinical presentations, including monophasic or recurrent episodes of optic neuritis, transverse myelitis, brainstem and cerebellar demyelinating attacks, monofocal or polyfocal cerebral deficit, and cerebral cortical encephalitis, with associated seizures.2 The average age at onset is approximately 30 years, and around 30% of cases are in the paediatric age group.1
Following an acute attack, most patients with MOGAD experience rapid improvement, to full or near normal visual acuity after corticosteroid therapy.2 Some have poor recovery with long-term damage to bladder, bowel, or sexual function.2 Patients who are younger at the time of initial presentation are at higher risk for relapse, versus those with presence of isolated transverse myelitis or acute disseminated encephalomyelitis (ADEM)-like presentation, who are likely to have a lower relapse risk.3
MOGAD has many overlapping clinical features with conditions such as MS and NMOSD,2 which require very different management approaches, making diagnosis challenging. In recent years, the increased availability of CBAs for the detection of MOG-IgG has allowed clinicians to distinguish MOGAD as a separate entity from MS and NMOSDs, including aquaporin-4 (AQP4) NMOSD.2 However, it is important to consider the limitations of these tests, and to use their results alongside those of clinical presentation and neuroimaging. A proposed expert consensus diagnostic pathway, published in March 2023, outlined a stepwise approach that starts with a set of core demyelinating events, which should prompt MOG-IgG testing, and explained when additional clinical or MRI features are necessary to confirm a diagnosis (Table 1).2
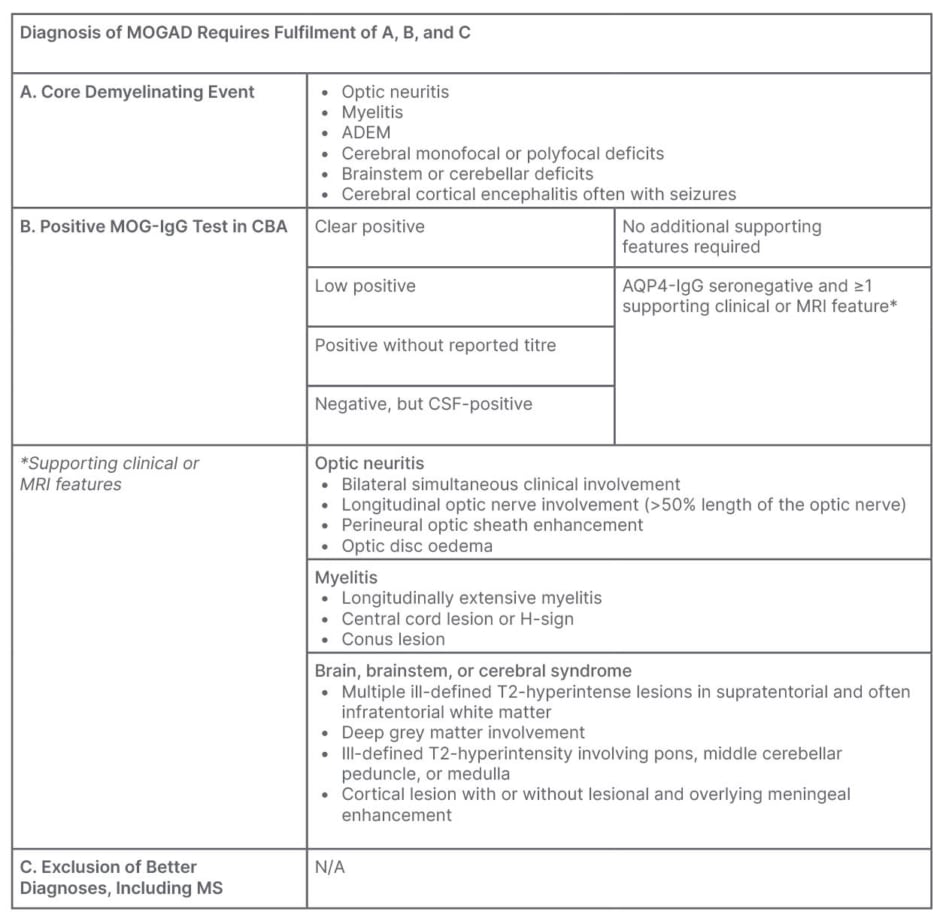
Table 1: Proposed diagnostic criteria for MOGAD.2
ADEM: acute disseminated encephalomyelitis; AQP4: aquaporin-4; CSF: cerebrospinal fluid; CBA: cell-based assay; MOG: myelin oligodendrocyte glycoprotein; MOGAD: MOG antibody-associated disease; MS: multiple sclerosis; N/A: not applicable.
Pathology and Clinical Presentation
The clinical signs of MOGAD include optic neuritis, myelitis, ADEM, cerebral monofocal or polyfocal deficits, and cerebral cortical encephalitis, often with seizures,2 though these often vary with age. ADEM, for example, occurs more frequently in paediatric than adult patients, whereas the reverse is true for optic neuritis, which is the most common symptom in adults.2 People presenting with optic neuritis, which is often bilateral,2,4 frequently experience severe vision loss and pain.5 Myelitis occurs in up to 30% of adults with MOGAD, and about 11% of children,6 and, while it may be severe at nadir, motor and sensory recovery is usually excellent.2 Brainstem or cerebellar involvement, which can cause dysarthria, is present in approximately 30% of patients.7,8 Seizures, which may be the presenting feature, particularly in children, tend to be focal motor or focal to bilateral tonic-clonic, and are usually self-limiting.9
In patients with suspected MOGAD, testing for serum MOG-IgG can be useful, but the positive predictive value varies according to the patient population.10 MOG-IgG antibodies are, for example, present more often in paediatric (40%) than in mixed (29%) and adult (22%) acquired demyelinating syndrome cohorts.11 They also occur more often in those who present with ADEM or optic neuritis. As such, the results should only be used alongside clinical characterisation and neuroimaging.10
In addition, a lack of standardisation to testing approaches can impact on reproducibility and accuracy of MOG-IgG results, particularly at low titres.12 In a bid to overcome such concerns, the authors of the recently-published proposed diagnostic criteria endorsed the use of CBAs. While live CBAs, quantified by flow cytometry or microscopy, offer higher sensitivity and specificity than fixed CBAs, the latter are a reasonable alternative when the former are unavailable.2 Enzyme-linked immunosorbent assays were not recommended.2
Serum Versus Cerebrospinal Fluid for Diagnosis
While the first consensus document from an international expert panel for the diagnosis and management of MOGAD, published in 2019, recommended measurement of MOG antibodies in serum only,13 the 2023 expert opinion advises cerebrospinal fluid (CSF) screening in some cases, for instance in seronegative patients with clinical and MRI characteristics of MOGAD.2 In patients who are subsequently CSF positive, the presence of at least one additional supportive clinical or MRI feature is necessary to make the diagnosis.2
This position, the speakers explained, follows studies that have identified patients with MOGAD with antibodies in both serum and CSF, and in the CSF alone, and associated these cases with compatible clinical/pathological phenotypes.14,15 Multicentre studies have also described associations between CSF positivity and a clinical phenotype compatible with myelitis/encephalitis, and more severe disease.16,17
While the speakers believed there are still several open questions related to CSF MOG antibody testing, including a lack of cut-off values and the need for further validation, they said it may have the potential to increase diagnostic sensitivity and identify patients with more severe disease.
Integrating Imaging into MOGAD Diagnosis
Imaging is an important tool for supporting diagnosis, yet there can be some overlap between MOGAD and other demyelinating diseases. Between 14.3–26.9% of patients with MOGAD, for example, meet the Barkhof MRI criteria for a diagnosis of MS.18 It is crucial, then, to understand the features that differentiate such conditions from each other, and how to integrate imaging results into diagnostic pathways.
Key characteristics for differentiating MOGAD from AQP4-NMOSD or MS include:
• Brain imaging may be normal in patients with MOGAD with optic neuritis or transverse myelitis, while those with MS will have multifocal T2-hyperintense white matter lesions.2
• The clinical course in MOGAD is monophasic or relapsing, while in AQP4-NMOSD and MS, it is predominantly relapsing.2
• Oligoclonal bands are present in CSF, but not serum, in 5–20% of those with MOGAD, and >80% of those with MS.2
• In MOGAD, there is demyelination with perivenous accentuation that involves both complement and CD4+ T cells. In MS, demyelination is associated with axonal damage, and involves astrogliosis and CD8+ T cells.19,20
Key Differentiations on Imaging
Distribution of brain lesions on MRI has been shown to differentiate MOGAD from MS with 91% sensitivity and 95% specificity.21 One cross-sectional study compared brain and spinal cord scans from 116 patients with AQP4-NMOSD in the chronic disease phase to 65 age-, sex-, and disease duration-matched patients with MS. It indicated that fulfilling at least two of five criteria (absence of juxtacortical/cortical lesions; absence of periventricular lesions; absence of Dawson fingers; presence of long transverse myelitis; presence of peri-ependymal lesions along the lateral ventricles) discriminated patients with NMOSD from those with MS.22
While much work is ongoing in this area, the recently published diagnostic consensus criteria for MOGAD have summarised MRI features that differentiate it from both AQP4-NMOSD and MS.2 MRI features supportive of a diagnosis of MOGAD, it states, include fluffy or poorly demarcated T2 lesions, the presence of lesions in the white matter, deep grey matter, middle cerebellar peduncle, large lesions in the brainstem, and confluent lesions in the cerebral cortex. There will also be non-specific leptomeningeal enhancement around the brainstem, and unilateral or bilateral cortical linear leptomeningeal enhancement, with cerebral cortical encephalitis, while residual T1-hypointense lesions are extremely rare. Lesions characteristic of MS, i.e., small, ovoid T2-hyperintense lesions in the juxtacortical and periventricular white matter, and persistent T1-hypointense lesions, are uncommon in MOGAD.2
Evaluation of optic neuritis is also key to differentiating MOGAD from other aetiologies. MOGAD optic neuritis is more commonly bilateral than that caused by MS.2 In MOGAD, attacks are often severe,2 but usually respond to corticosteroid treatment, especially if given early.23 MOGAD optic neuritis is associated with optic disk oedema and perineural optic nerve enhancement.2
Paediatric Versus Adult MOGAD Imaging Features
Both similarities and differences in the presentations of adults and paediatric patients with MOGAD exist. Monophasic ADEM, for example, is much more common in children than in adults,14 and paediatric patients also have a wide range of differential diagnoses, including other antibody-mediated and neurometabolic conditions, tumours, and macrophage activation syndrome.2
MRI imaging in paediatric patients with autoimmune encephalitis and MOG-IgG antibodies has indicated cortical and deep grey matter involvement in all children, and, in 60%, juxtacortical signal alterations. There was no involvement of other white matter structures or contrast enhancement.24 Furthermore, comparison of the clinical and neuroradiological features of paediatric patients with ADEM, with and without MOG-IgG antibodies, has indicated a nearly uniform MRI pattern, characterised by large, hazy, and bilateral lesions, and the absence of atypical MRI features (e.g., mainly small lesions, well-defined lesions), with involvement of more anatomical areas, which was significantly different compared to that of children without MOG antibodies.25
Additional features that help differentiate MOGAD from AQ4P-NMOSD and MS include the occurrence of ADEM, monophasic course of disease off treatment, risk for residual sphincter and erectile impairment despite good motor recovery from a transverse myelitis, moderate-to-severe oedema, and haemorrhage of the optic nerve head, as well as extremely rare occurrence of residual T1-hypointense lesions.2
Red flags indicative of a diagnosis other than MOGAD include progressive neurological impairment in the absence of attacks, lack of efficacy for high-dose corticosteroids in acute attacks, fulfilment of the MS MRI criteria, silent lesions outside of relapse, and lesion contrast enhancement that persists for ≥6 months.2
Current Management
There is currently no approved treatment for MOGAD. The speakers explained that management has three main aims: resolving acute attacks, preventing early rebound attacks, and the long-term management of disease. Prompt intervention with an intravenous steroid after an acute attack significantly improves both visual and structural (peripapillary retinal nerve fibre layer) outcomes in patients with MOGAD.23,26 Other options for treatment for acute attacks are intravenous immunoglobulins and plasma exchange. A short follow-on treatment course, with an agent such as prednisolone, can reduce the risk of early relapse.3 A number of approaches can be employed for long-term relapse prevention. These include monthly maintenance intravenous immunoglobulins, conventional immunosuppressants, and rituximab.27
MOGAD and Pregnancy
MOGAD affects females of childbearing age, meaning patients may require acute or long-term interventions prior to, during, or post-pregnancy.28 Addressing pregnancy in females living with MOGAD helps to guide disease management and the intensification of monitoring, thereby potentially decreasing the risk of complications.28 In addition, answering their pregnancy-related questions can help females to make informed decisions regarding family planning,28 and reduce their anxiety. A few retrospective studies that enable healthcare professionals to have these conversations have been published in recent years.
One study investigated the influence of pregnancy on patients with NMOSD or MOGAD, with the aim of identifying factors that predicted pregnancy-related attacks.29 Twenty-one patients who were MOG antibody-positive experienced a total of 28 pregnancy-related attacks, 11 of which occurred during the first trimester post-partum. Pregnancy outcomes included 18 term deliveries, one premature delivery, and two elective abortions. No spontaneous abortion, neonatal malformations, or pre-eclampsia were observed.29
Another study included 30 pregnancies in 20 patients with MOG antibodies. There was no significant increase in the annualised relapse rate for any trimester during pregnancy or the post-partum period, when compared to pre-pregnancy annualised relapse rate. Pregnancy outcomes were unremarkable in a majority of the patients.30
A third analysis included results from 20 females (25 pregnancies) with MOGAD who became pregnant after disease onset. No relapse was recorded during the pregnancies, and only three relapses occurred during the first 3 months post-partum. The annualised relapse rate decreased from 0.67 during the period prior to pregnancy to 0.00 during pregnancy, and to 0.22 during the first year post-partum.31
Together, such results suggest a reduction of MOGAD relapses during pregnancy, and a potential modest impact during the first 3 months following delivery. Outcomes for mothers with MOGAD and their offspring appear to be within the range reported for those without the disease. Nevertheless, careful treatment considerations should be discussed in advance, and multidisciplinary specialised follow-up, for both the mother and the child, arranged.
Future Directions
Recent years have witnessed a significant evolution in the number of diagnostic, prognostic, and therapeutic biomarkers for distinguishing MOGAD from NMOSD and MS, opening up the possibility of novel therapeutic strategies.
Levels of multiple cytokines, including interferon-gamma, IL-13, IL-6, IL-8, granulocyte-monocyte colony-stimulating factor, IL-1 receptor antagonist, monocyte chemotactic protein-1, and macrophage inflammatory protein-1α, have been shown to be higher in patients with MOGAD or NMOSD, than in those with MS, for example.32
Human endogenous retrovirus-W may also be a prognostic biomarker in neuro-inflammatory disease. Antibody reactivity against human endogenous retrovirus-W envelope peptides was similar in MOGAD and MS, and differed from those with AQP4-NMOSD.33 Glial fibrillary acidic protein, a marker of astrocyte damage, and neurofilament light chain, a marker of neuroaxonal damage,34 are both elevated in patients with either MS or NMOSD versus healthy controls, and levels are higher during relapses than remission.35 Complement component 3 and 4 levels are elevated in patients with MOGAD compared to both healthy controls and patients with NMOSD,36 and complement activation has been shown to be a prominent feature of MOGAD.37
There has also been interest in novel biomarkers. A multicentre study of patients with demyelinating disorders who were double seronegative for AQP4 and MOG-IgG antibodies indicated that some were positive for MOG-IgA.38 These patients presented with frequent myelitis and brainstem syndrome, infrequent optic nerve involvement, and a low percentage of CSF-specific oligoclonal bands.38 It suggests that MOG-IgA may be a novel diagnostic biomarker for a distinct subgroup of double-seronegative patients with CNS demyelination.
Based on such findings, the number of agents that may become available for relapse prevention is increasing. Theoretically, newer agents that target CD19, the IL-6 receptor, and the neonatal Fc receptor also may prove effective,27,39 and Phase III trials to assess their safety and efficacy are ongoing.
More information is also being accumulated about what does not work. There now is evidence that agents used to treat MS are not effective in MOGAD, and that rituximab is less effective in MOGAD than in AQP4-NMOSD.40-42 Complement activation appears to play less of a role in MOGAD pathogenesis than in that of AQP4-NMOSD, suggesting that treatments targeting this pathway may not prove useful in MOGAD. However, more clinical studies in this area are needed.43
MOG-IgG Titres
There has been some debate around the value of assessing MOG-IgG titres and persistent positivity during follow-up. It has been suggested that the temporal dynamics of MOG-IgG may inform diagnosis, help to determine the risk of relapse and disability, and inform treatment decisions.44,45 However, challenges in the monitoring of MOG-IgG dynamics remain.
Firstly, MOGAD treatment may influence results of MOG antibody testing. In theory, plasma exchange may lead to false negative results, the administration of intravenous immunoglobulins may decrease the signal-to-noise ratio or lead to false positives, and B cell depletion with rituximab may decrease MOG-IgG detection, though data in this area are currently lacking. There is also a lack of standardisation across laboratories, and limited reliability for the quantification of low titres.2 Other questions, the speakers explained, include debates over whether serum or CSF samples should be utilised, the most appropriate type of assay (live versus fixed cell-based) to use, the characteristics of secondary antibodies, validation of cut-off values, and whether variations in patient-history influenced follow-up in clinical practice.
They also argued that it was important to consider the probability and importance of events that might be captured by monitoring titres. Persistent MOG-IgG positivity has been shown to be associated with higher frequencies of a relapsing clinical course in children with MOGAD,46 and seroreversion has also been associated with substantial reductions in relapse risk.47 However, some patients relapse despite being MOG-IgG negative, and seroreversion may equate to the antibody level dropping below a cut-off threshold, rather than disappearing completely.48 In addition, there is significant cost and patient inconvenience associated with repeated antibody assessments in clinical practice.48
The speakers argued that monitoring and acting on information provided by MOG-IgG titres requires better standardisation of assays, and more prospective studies assessing MOG-IgG temporal dynamics over long-term follow-up. If this approach to attack prediction is to be advanced, alternative, less burdensome methods of specimen collection, such as at-home blood spot, tears, or urine collection, may need to be considered. The assessment of additional antibody isotypes may also need to be incorporated to provide additional insights.
Conclusion
In conclusion, the masterclass speakers explained that the clinical characteristics of demyelinating conditions, such as MOGAD, MS, and NMOSD can overlap, and that correct diagnosis is key to guiding the effective treatment of disease. Differentiation requires a holistic approach that combines patient history with the results of MOG antibody and neuroimaging tests, as well as the exclusion of differential diagnosis.






