
Abstract
This article describes a case of Turner syndrome (TS) associated with craniosynostosis due to an early closure of the sagittal suture. Skeletal anomalies are characteristic phenotypic findings, although the presence of associated craniosynostosis constitutes an unusual pathology with very few references in the literature. The case describes a 5-year-old patient who presented with failure to thrive, psychomotor retardation, and scaphocephaly. Genetic analysis showed an Xq isochromosome. Different genes have been identified in the pathogenesis of TS. The SHOX (short-stature homebox) gene and its interaction with other growth regulator genes is responsible for different bone anomalies in TS and in other skeletal dysplasias. Classic cephalometric studies have demonstrated marked alterations in the skull base in patients with TS. The association of abnormal cranial morphology together with the craniosynostosis could cause a decrease of volume in the posterior fossa. In this patient, the dynamic study of cerebrospinal fluid in flow MRI was normal; therefore, clinical, radiological, and ophthalmological follow-up was prescribed. Craniosynostosis is a rare entity in TS. The presence of premature closure of the skull sutures makes it necessary to rule out other abnormalities of the craniocervical junction.
INTRODUCTION
Turner syndrome (TS) is one of the most frequent chromosomal aberrations, caused by the partial or complete loss of one X chromosome.1 The disorder leads to a conglomerate of phenotypical manifestations that often includes short stature, congenital lymphoedema, gonadal dysgenesis, or cardiovascular and renal congenital malformations.2 Skeletal anomalies are a characteristic phenotypic finding, including disproportion between the upper and the lower body segments, cubitus valgus, scoliosis, genu valgum, short metacarpals, or congenital hip dislocation. Patients with TS may also have a variety of typical craniofacial manifestations such as sphinx facies, ogival palate, micrognathia, and short neck due to hypoplasia of cervical vertebrae or pterigium colli.3
However, the presence of associated craniosynostosis constitutes an infrequent entity, with few clinical cases reported in the literature. In 1959, the first case of a patient with TS and cranial malformation was reported in the shape of turricephaly due to premature closure of the coronal and sagittal sutures; the turricephaly was of mild degree and no surgery was required.4 In 1968, Calmettes et al.5 reported a case of oxycephaly and corneal dystrophy in a patient with TS because of its unusual combination. Another case was described by Bozzola et al.6 in 1986: their patient presented a complex craniosynostosis with the fusion of sagittal and bicoronal sutures; two surgical procedures were necessary to correct it.6 One year later, Massa and Vanderschueren-Lodeweyckx7 published the first case of monosutural craniosynostosis associated to TS. Radiological examination of the skull revealed a premature fusion of the sagittal suture and digital impressions on the parietal bones, although surgical management was not required.7 In a recent study that focused on the incidence of systemic diseases and syndromic diagnoses in a cohort of patients with scaphocephaly, a single case of TS was identified.8
TS and craniosynostosis is an unusual concurrence. However, with the current evidence, simple or severe forms of craniosynostosis should be considered as a possible skeletal abnormality in TS. In addition, molecular researchers have identified some genes that could be involved in the different clinical expressions of this pathology.2 This article describes a patient diagnosed with TS who presented an isolated sagittal synostosis. Likewise, morphological aspects in the development of the skull in the context of untreated craniosynostosis are reviewed and the possible genetic implications in this patient with TS is analysed.
CASE REPORT
History and Examination
The patient was a 5-year-old female, referred to the authors’ department one year prior due to cranial deformity including scaphocephaly. The cephalic index was 70. Antenatal history was unremarkable and delivery was uncomplicated. There was no parental consanguinity. Clinical examination showed a decrease in growth velocity for her age; at 6 years old she weighed 14.40 kg and her height was 108 cm. No other remarkable musculoskeletal characteristics were identified. Mild developmental concerns were identified in visuospatial skills, non-verbal perceptual problems, and attention disabilities.
Complementary Examinations
Funduscopy was consistent with papillary paleness and visual evoked potentials and showed a very mild delay through the optic pathways. A CT scan revealed a craniosynostosis due to premature closure of the sagittal suture (Figure 1). MRI of the brain and cervical region showed a minor descent of the cerebellar tonsils through the foramen magnum, at the lower limit of normality.
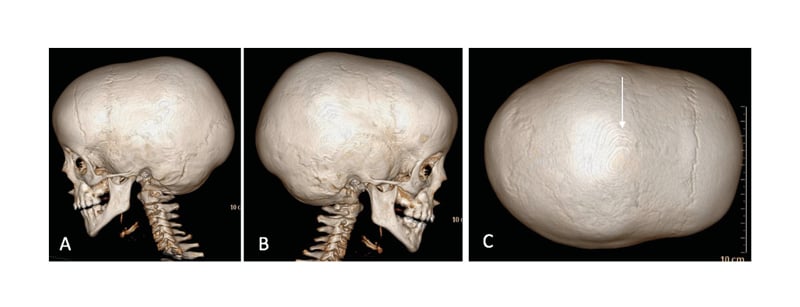
Figure 1: CT scan with 3D reconstruction showing: A–B) lateral view; C: vertex view.
The arrow shows a synostosis sagittal suture.
No other abnormalities were found in the morphology or signal of the brain parenchyma (Figure 2A, B, and D). The dynamic study of the cerebrospinal fluid (CSF) in flow MRI did not reveal compromise of CSF flow at the occipitocervical junction level (Figure 2C).
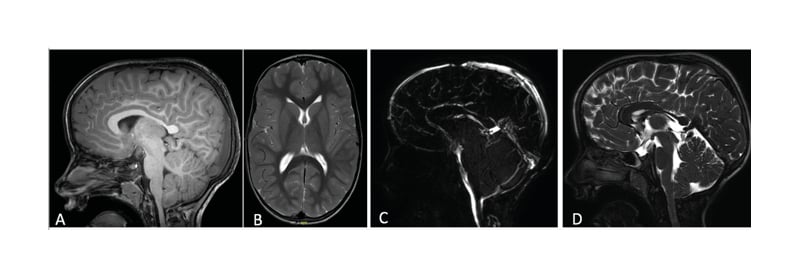
Figure 2: A–B) T1 sagittal and T2-weighted axial magnetic resonance brain images show a cranial deformity in the context of an early closure of the sagittal suture. There are no notable changes in the morphology and signal of the brain parenchyma; C) dynamic study of cerebrospinal fluid in flow MRI is normal, without any compromise of cerebrospinal fluid flow; D) T2-weighted sagittal magnetic resonance brain image demonstrates minor descent of the cerebellar tonsils through the foramen magnum at the lower limit of normality.
Cardiovascular health issues were normal. Screening renal ultrasonography showed no renal malformations. Laboratory tests of renal function were normal. The patient initiated treatment with growth hormone due to short stature coupled with an insulin like-growth factor 1 value at the lower limit of normality (65 ng/mL) and a bone age ahead of 1 year, which caused a prognostic height below her target size. Neuropsychological and behavioural monitoring is ongoing due to developmental delays.
Conventional karyotyping showed a nonmosaic 45,X,i (X)(q10). In all metaphases (20), the presence of two X chromosomes, a structurally normal X, and an Xq isochromosome were observed, constituting a partial Xq trisomy and a partial Xp monosomy. Fluorescence in situ hybridisation probes showed two X chromosomes, one of them with a normal hybridisation pattern for both subtelomeric regions (one signal for Xpter/one signal for Xqter) and the second chromosome with an altered hybridisation pattern for the subtelomeric regions of the short and long arms of the X chromosome. No hybridisation signal was observed for the SRY region. Karyogram and fluorescence in situ hybridisation images are described in Figure 3. Array comparative genomic hybridisation analysis confirmed the existence of a probably terminal partial deletion of 53 Mb in the short arm of the X chromosome, as well as the presence of a partial duplication of 85 Mb of the long arm of the X chromosome, involving chromosomal bands Xq13–q28. Both of them altered the dose of several reference genes. The imbalances identified were compatible with the presence of a derivative X chromosome.
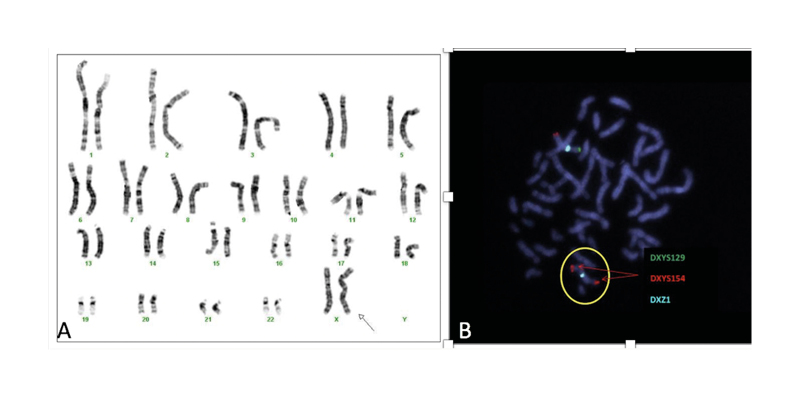
Figure 3: A) Karyotype of the patient, a non-mosaic 45,X,i (X)(q10); B) fluorescence in situ hybridisation showing a metaphase with two X chromosomes (DXZ1x2), one of them with a signal for each of the hybridised subtelomeric regions (DXYS129 +, DXYS154 +; pattern compatible with normal X chromosome) and the second chromosome (circled) with two signals for the subtelomeric region of the long arm of the X chromosome and no signal for the subtelomeric region of the short arm of the X chromosome (DXYS129, DXYS154 ++; pattern compatible with an X chromosome with qter duplication and pter deletion).
DISCUSSION
TS is a well-known chromosomal disorder caused by a partial or complete lack of one of the X chromosomes. The karyotype variation is large and complex.1 The relationship between phenotype and genotype is being widely investigated. Clinical manifestations are not only related to the grade of deficit of X chromosomal material, but also depends on the expression of different genes and epigenetic and transcriptional factors.9 The SHOX gene is situated in pseudoautosomal region 1, a chromosomal segment located on the short arm of the X and Y chromosomes (Xp22.3 and Yp11.3). This gene, a transcriptional regulator, is involved at the point of fusion of the growth plate and skeletal maturation.10 The function of this gene is dose dependent and is associated with short stature and other skeletal anomalies.9 In TS, the haploinsufficiency of the SHOX gene, located on the short arm of the X chromosome, is justified by the presence of isochromosomes from the long arms of the X chromosome. In fact, growth deficit tends to be more pronounced in X ring chromosome and isochromosome Xq karyotypes than 45X.11 The karyotype of the patient was nonmosaic 45,X,i (X)(q10) and she had a short stature. Furthermore, preclinical investigations have identified a complex interaction between SHOX gene and other growth regulator genes such as fibroblast growth factor receptor 3, offering a possible theory about the different bone abnormalities in TS, as well as in other skeletal dysplasia (e.g., achondroplasia).12,13
Likewise, classic cephalometric studies have demonstrated marked alterations in the skull base in patients with TS. The clivus is shorter and the angle between the sphenoidal plane and the Sella-Nasion line is larger, similar to the angle between the foraminal and clival planes; however, measurements of anterior cranial fossa are normal.14 Further research is needed to determine the genetic mechanisms behind craniosynostosis such as skull morphology in TS. In any case, the association of an abnormal cranial morphology together with the presence of scaphocephaly could cause a decrease in volume in the posterior fossa. Chiari malformation Type 1 has been described as a possible complication of untreated sagittal synostosis.15 The early closure of the sagittal suture produces an increase in the skull in the axial plane and a restriction of the vertical growth of the cranial vault, resulting in a posterior fossa smaller than normal. Decreased supratentorial volume leads to the descent of the cerebellar tonsils, either directly or by intracranial hypertension associated with increased venous pressure due to constriction of superior sagittal sinus in the bone groove.15,16
Thus, in any case similar to the patient with a diagnosis of TS and the occurrence of premature closure of the cranial vault sutures, a high suspicion of associated craniocervical malformations must be considered. In this patient, the dynamic study of CSF in flow MRI was normal and ophthalmological control did not detect papilledema, so clinical, radiological, and ophthalmological follow-up was prescribed.






