
Abstract
Neurodegenerative disorders constitute a worldwide concern attributable to the ageing of the human population. In this context, Alzheimer’s disease (AD) accounts for up to 70% of dementia cases worldwide. With no effective treatment available, new therapeutic alternatives are under assessment. Recently, several researchers have highlighted the need to include the multifactorial aetiology of AD as part of the design and evaluation of novel AD-therapies. Thus, it is not enough to only understand the critical molecular events that occur during AD pathophysiology, but to unveil the crosstalk between these events as well as its interplay with different biological subsystems. This is the case for neuroinflammation, an extremely complex response, widely recognised as a main contributor of AD-linked neurodegeneration but poorly understood in terms of its physiological interactions. Accordingly, based on previous work regarding the relationship between the Wnt signalling pathway and toll-like receptor-mediated inflammatory response, this review provides an update to the integrative view of this communication and discusses future directions of research focussed on modulating the inflammatory response within the central nervous system of AD patients.
INTRODUCTION
Described in 1906, Alzheimer’s disease (AD) has become a major burden to the human population. Highly correlated with ageing, it has been estimated that AD can account for up to 70% of dementia cases worldwide with a cost of over $800 billion to public health systems around the world.1 Accordingly, substantial human and financial efforts have been committed to fight AD. Regrettably, even with our increased understanding of the molecular mechanisms of the disease, no success has been achieved in the different clinical trials developed during the last decade.2 In this regard, there is an urgent need to reconsider the way we approach potential AD therapeutics, such as the reappraisal of already known drugs, but taking the multifactorial aetiology of the pathology into consideration. Notably, during the last years it has become evident that addressing the inflammatory process during AD is fundamental not only to properly understand the pathology but to depict future therapies.
ALZHEIMER’S DISEASE
AD is commonly associated with a progressive loss of memory; however, the disease usually begins earlier with almost imperceptible symptoms such as subtle abnormal social behaviours or mood changes. Therefore, memory alterations only become evident with the progression of the disease and when the underlying neuronal dysfunction is enough to alter brain function.3-7 Although the accumulation of amyloid-β (Aβ) peptides and hyperphosphorylated tau aggregates have been defined as the molecular hallmarks of the disease,7-9 it is currently widely accepted that additional pathological features of the disease, such as vascular alterations,10 mitochondrial dysfunction,11 and neuroinflammation,12 are involved from the very beginning in the progression of the disorder and appear even earlier than the traditional hallmarks.
Alzheimer Amyloid-β Peptide
According to the amyloid hypothesis of AD,6 Aβ is a pivotal factor regulating the aforementioned features including the neurofibrillary tangles. Indeed, our current understanding of the effects of Aβ on the cellular molecular system has improved significantly during the last decades.13-15 Oxidative stress, mitochondrial alterations, hyperphosphorylation of tau protein, further Aβ production, synaptic disruption, and neuronal cell death have been linked with the direct effects of Aβ on different organelles and/or molecular cascades.16,17
In this regard, Aβ constitutes a 37–43 amino acids post-transcriptional product of the amyloid precursor protein (APP). APP processing involves two possible pathways: 1) The non-amyloidogenic processing is carried out by the α and γ secretases, leading to the release of the soluble APPα and p3 fragment; 2) the amyloidogenic pathway is carried out by the β and γ secretases, leading to the release of the soluble APPβ and the neurotoxic Aβ peptide.18,19 Even when Aβ aggregates localise outside the cells they can also accumulate inside the neurons,20 being found within the mitochondria pool.21 Considering that APP is synthesised and processed within different subcellular compartments, finding Aβ within these structures is still possible. Moreover, it has been demonstrated that cells can uptake Aβ from the extracellular space through the α7-nicotinic acetylcholine receptor further supporting the intracellular accumulation of the peptide and that this can cause primary cellular alterations, including tau hyperphosphorylation and neurite atrophy.18 Importantly, these features are the most relevant regarding the neuroinflammatory cascade triggered during AD pathophysiology.
AΒ-DRIVEN NEUROINFLAMMATION AND THE ROLE OF TOLL-LIKE RECEPTORS
During the last decades it has become evident that sustained exposure to Aβ will lead to a chronic inflammatory state which ultimately will alter the brain microenvironment causing neuronal damage and/or neuronal death.22,23 Aβ causes increased levels of several proinflammatory mediators including various members of the IL family (IL-1β, IL-6, IL-12), TNFα, COX2, and inducible nitric oxide synthase.24-26 On the other hand, Aβ induces reactive oxygen species production through direct interaction with the mitochondria, not only affecting the metabolism of ATP but also leading to further production of proinflammatory mediators through NF-κB activation.27-31
These inflammatory mechanisms are exerted mostly via Aβ/toll-like receptor (TLR) interactions.26
The Brain and Toll-Like Receptors
Neurons are highly specialised cells with specific microenvironmental requirements. Accordingly, the central nervous system (CNS) remains a partially isolated domain with critical structures, such as the blood–brain barrier and the choroid plexus, and non-neuronal cells playing a fundamental role to sustain neuronal activity and protect the CNS from damage.22,26,32 Astrocytes, oligodendrocytes, and microglia are recognised as the support cells for neuronal activity, with astrocytes and microglia as the main effectors of the response against inflammatory-related pathological processes in AD.33 It is important to note that microglia remain the only representatives of the immune lineage within the CNS attributable to the colonisation of the brain by macrophages early during development.34
Beyond the different functions carried out by these cell types, each of them, including neurons, expresses different members of the TLR family. Thus, neuronal and non-neuronal cells cannot only respond to damaging insults but will also be affected by the presence of proinflammatory mediators.
The TLR family constitutes the main type of pattern recognition receptors which recognise the pathogen-associated molecular patterns and the damage-associated molecular patterns.35,36 Up to 13 members have been described for the TLR family with different localisation within the cells. Relevantly, TLR1,2, and 4–6 are present in the plasma membrane, usually sensing or interacting with pathogen-associated molecular patterns; while TLR3 and 7–9 localise to endosomes and are able to sense damage-associated molecular patterns including ATP and nucleic acids.36
As mentioned previously, neurons and glial cells express TLR but with a different pattern. While the microglia and neurons express all TLR subtypes, astrocytes express TLR2–4, 9, and 11.37,38
The canonical molecular cascade linked to the activation of the TLR have been revised elsewhere; however, Figure 1A summarises the main events derived from such activation. For the purpose of this revision it is relevant to highlight that TLR activation will ultimately lead to increased release of proinflammatory mediators including IL, TNF, transforming growth factor, IFN, and complement proteins, among others.39,40 In this context, it has been well established that Aβ triggers the inflammatory response mainly through direct interaction with TLR2 and TLR4, although it can interact with additional members of the TLR family.21,40-43 Consequently, the permanent exposure to high levels of Aβ will result in the activation of the TLR in neuronal and non-neuronal cells causing a chronic condition with a vicious circle of activation of the inflammatory cascade. According to this information, it is not surprising that anti-inflammatory therapies are proposed to be re-evaluated because of their potential to control the inflammatory component of the disease. Therefore, one element that should not be overlooked during this approach is the crosstalk between the inflammatory cascade and critical signalling pathways for neuronal physiology, such as the Wnt pathway.
WNT SIGNALLING/TOLL-LIKE RECEPTORS AND THE INFLAMMATORY MILIEU
The Wnt pathway constitutes a complex cellular signalling system which has been related to cell proliferation and differentiation.44,45 Wnt signalling can be divided in the canonical and the non-canonical Wnt pathways (Figure 1B). In the canonical pathway, Wnt proteins bind to the Frizzled receptor and low-density lipoprotein receptor-related protein 5/6, leading to the activation of dishevelled phosphoproteins and the interaction of low-density lipoprotein receptor-related protein 5/6 with axin. These events will cause the disassembly of the β-catenin destruction complex (adenomatous polyposis coli, axin, glycogen synthase kinase 3 beta [GSK-β3], and casein kinase 1), preventing the GSK-3β-mediated β-catenin phosphorylation. Then, the stabilised β-catenin can translocate to the nucleus where it will induce the expression of the Wnt target genes by binding to the T-cell factor and lymphoid enhancer-binding factor. The absence of Wnt ligands will define the turn-off of the system, causing the stabilisation of the destruction complex and the full activity of the GSK-3β, leading to β-catenin destruction.46,47 On the other hand, the non-canonical Wnt pathway, which can be divided in the Wnt/planar cell polarity and the Wnt/Ca2+ pathway, will lead to the activation of the c-Jun N-terminal kinase activity and to the rearrangement of cytoskeletal proteins,48,49 and to the activation of calcium-related proteins such as protein kinase C and calcium/calmodulin-dependent protein kinase II.48,49 Based on this division, it is possible to recognise two main types of Wnt ligands: those that activate the canonical pathway, including Wnt-1–3, 3a, and 8a; and those that are able to activate the non-canonical pathways, including Wnt-4, 5a, 5b, 6, 7a, and 11. However, this classification is not completely accurate since different ligands are able to activate depending on the physiological context, one way or another.50 Furthermore, several elements of the Wnt cascade have been described as master switches that can be accessed through additional signalling pathways. Interestingly, the inflammatory master NF-κB pathway is one of the molecular cascades able to interact directly with Wnt signalling.16
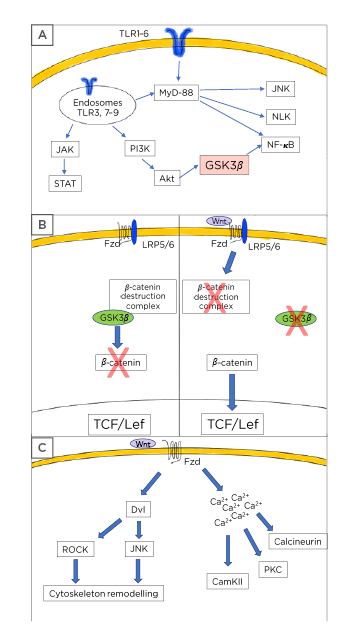
Figure 1: Toll-like receptor and Wnt pathway description.
A) Toll-like receptors (TLR) usually signal through the myeloid differentiation factor 88 leading to activation of the NF-κB pathway with the subsequent production and release of inflammatory mediators. Additionally, TLR also causes Nemo-like kinase and c-Jun N-terminal kinases activation. Some TLR can signal via the PI3K/protein kinase B/glycogen synthase kinase 3 β axis to induce further NF-κB activation. A third mechanism of TLR activity includes the JAK/STAT activation. B) Wnt signalling is composed of two main pathways. In the canonical, or Wnt/β-catenin dependent mechanism, the presence of Wnt ligands causes the activation of the Frizzled (Fzd)/low-density lipoprotein receptor-related protein 5/6 receptor leading to the disassembly of the β-catenin destruction complex and preventing the GSK-3β-mediated β-catenin phosphorylation. Thus, stabilised β-catenin can translocate to the nucleus and bind to the T-cell factor/lymphoid enhancer-binding factor transcription factor allowing the transcription of the Wnt target genes. In the absence of Wnt ligands, the system is turned-off and GSK-3β can phosphorylate β-catenin leading to its proteasomal destruction. C) The Wnt pathway also considers the non-canonical cascade, in which the presence of specific Wnt ligands will cause cytoskeleton rearrangement through dishevelled phosphoproteins followed by Rho-associated coiled-coil containing protein kinase and c-Jun N-terminal kinases activation in the planar cell polarity pathway. Similarly, the non-canonical activation can trigger the Wnt/Ca2+ cascade leading to increased levels of calcium from intracellular storages and to the activation of several calcium dependent proteins, such as calcineurin, calcium calmodulin kinase II, and protein kinase C.
Akt: protein kinase B; CamKII: calcium calmodulin kinase II; JNK: c-Jun N-terminal kinases; Dvl: dishevelled; Fzd: Frizzled; GSK-3β: glycogen synthase kinase-3 β: Lef: lymphoid enhancer-binding factor; LRP: low-density lipoprotein receptor-related protein; MyD88: myeloid differentiation factor 88; NLK: Nemo-like kinase; PKC: protein kinase C; ROCK: Rho-associated coiled-coil containing protein kinase; Tcf: T-cell factor.
Wnt and Toll-Like Receptors Connection
In their previous work, the authors underlined the close relation and the reciprocal modulatory effect between TLR and Wnt signalling. Part of the molecular cascade triggered through TLR activation can be tracked down to the molecular switches present in the Wnt pathway. For example, TAK1, the controller of the IκB kinase complex, also activates the Nemo-like kinase and the c-Jun N-terminal kinase, both a factor and an end point of the activation of the canonical and non-canonical Wnt pathways.51-53 Similarly, the potential regulation of GSK-3β through the TLR-PI3K-protein kinase B axis also contributes to solve the tight relation observed between Wnt signalling and the inflammatory response.53 New research further supports this connection and underlines the critical role that Wnt signalling plays in the neuroinflammatory process.
In this context, the work of Song et al.54 and Van Steenwinckel et al.55 demonstrated not only the ability of Wnt signalling to modulate the activation of microglia, but to also downregulate the increase in the levels of critical proinflammatory mediators. Interestingly, the TWS119-induced inhibition of GSK-3β not only ensures canonical Wnt activity but also prevents the binding of NF-κB with the cyclic adenosine monophosphate response element binding protein-binding protein causing the blockade of the synthesis of several cytokines, a relevant issue beyond microglia activation. Indeed, TWS119 also acts as a regulator of the PI3K/protein kinase B/GSK-3β/reactive oxygen species axis.56 The use of specific Wnt ligands, such as Wnt3a, has demonstrated significant anti-inflammatory effects including inducible nitric oxide synthase and TNFα downregulation.56
The work of Royer et al.57 demonstrated the complementarity of the TLR/Wnt connection. In their work, the activation of TLR3 led to an increase in the levels of matrix metalloprotease 9 but in a mechanism absolutely dependent on the activation of the Wnt/β-catenin pathway. A similar complementary effect was also observed between the TLR4 and the Wnt/Dickkopf-related protein 3 axis.58 This constitutes a relevant mechanism in tumour growth regulation in which the downregulation of the TLR4 will increase the activity of the Wnt pathway.
On the other hand, it has recently been demonstrated that the activation of Wnt signalling is closely related with the neuroprotection necessary in spinal cord injury. Moreover, this activation can be linked with several processes including blockade of apoptosis, tissue repair, and modulation of the inflammatory response.59
It is well known that Wnt signalling can exert different functions depending on the cellular context. Relevantly, this situation can also be observed regarding the inflammatory modulation exerted by some Wnt components. In this regard, different works have demonstrated that β-catenin, the main effector of canonical Wnt signalling, can be associated with several inflammatory conditions including infections and sterile processes such as colitis, liver injury, and myocardial infarction.60-63 Furthermore, the work of Huang et al.64 has recently demonstrated that β-catenin leads to the activation of the NOD-, LRR-, and pyrin domain-containing protein 3 inflammasome, a critical regulator of cytokine production.64
CONCLUSION
The role that Wnt signalling plays in inflammation or as an immunomodulatory agent has been largely known in the context of cancer and other pathological conditions. Recent research has also pointed out that these effects might be of relevance during the neurodegenerative process involving inflammation, such as AD. Previously, the authors depicted a common line between TLR and the Wnt pathway and have here provided information regarding recent findings that further suggest that these two molecular cascades are closely related and that they are fundamental in understanding the complexity of the neuroinflammatory process observed during AD neurodegeneration.






