Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that is estimated to affect approximately 300,000 individuals worldwide. From symptom onset, the disease has rapid progression, and typically leads to death in approximately 3 years, though there is wide phenotypic variability. ALS pathophysiology is probably driven by several cellular and molecular mechanisms, including endoplasmic reticulum (ER) dysfunction, apoptosis, oxidative stress, impaired intracellular transport, neuroinflammation, and defective RNA metabolism and protein homeostasis. Several agents that target these pathways are in development, and a few are approved in certain regions. A fixed-dose combination of sodium phenylbutyrate and ursodoxicoltaurine (PB and TURSO, also known as AMX0035) was developed to target ER stress and mitochondrial dysfunction. This combination was approved for the treatment of ALS in the USA and Canada in 2022, following findings from the CENTAUR trial. CENTAUR was a Phase II trial comprising a 24-week randomised placebo-controlled phase and an open-label extension (OLE) phase. Treatment with PB and TURSO significantly slowed the rate of functional decline over 24 weeks compared with placebo, meeting the primary endpoint of the study. Over long-term follow-up, median survival duration was about 4.8 months longer in the group originally randomised to PB and TURSO, compared with the group originally randomised to placebo. PHOENIX, a Phase III trial of PB and TURSO planned to be completed in 2024, includes a 48-week randomised controlled phase, followed by an OLE. The PHOENIX trial is expected to provide additional insights regarding the effects of PB and TURSO in ALS.
INTRODUCTION
ALS is a neurodegenerative disease characterised by dysfunction and degeneration of the motor neurons, leading to progressive muscle weakness and atrophy. Globally, prevalence estimates for ALS range from about 1 to about 10 per 100,000 people, dependent on the country, with an estimated 300,000 people living with ALS worldwide (95% uncertainty interval: 213,893–310,663).1,2 Over time, people living with ALS lose the ability to move, swallow, speak, and breathe. Loss of independence and the need for assisted breathing and feeding often occur within the first 2−3 years, although the timeframe is highly variable.3-5 Respiratory failure eventually leads to death, often 3−5 years after onset.5-8 The disease can also impact cognitive function and quality of life, and may lead to caregiver and economic burdens.9-11
AMYOTROPHIC LATERAL SCLEROSIS PATHOPHYSIOLOGY
Neuropathologically, ALS is characterised by degeneration and loss of motor neurons in the motor cortex and anterior horns of the spinal cord. Affected motor neurons typically contain proteinaceous inclusions, and, in most cases, cytoplasmic inclusions contain tar DNA-binding protein 43 (TDP-43).7 Aggregated TDP-43 consists of truncated, polyubiquitinated, and phosphorylated protein.12-14 It has been proposed that mislocalisation of TDP-43 causes a loss of its normal nuclear function related to gene expression regulation at the pre-mRNA splicing level.15 Overall, neuronal degeneration in ALS is thought to be linked to many interplaying pathogenic pathways, several of which are summarised in Figure 1, and reviewed elsewhere.7,16,17 The complexity of ALS pathogenesis implies that therapeutic approaches capable of modifying multiple, preferably upstream, pathways of neurodegeneration may have the highest probability of successful clinical outcomes.
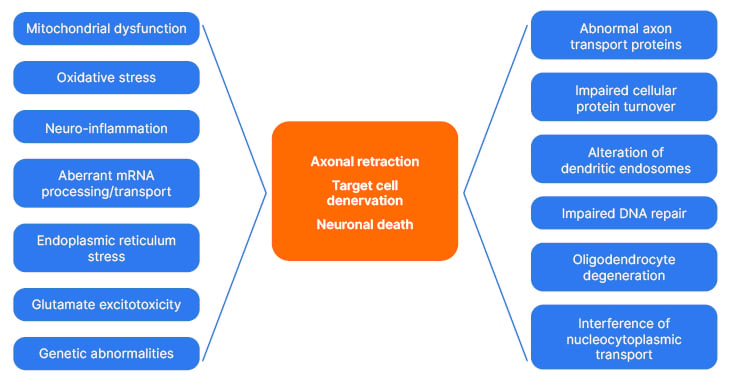
Figure 1: Potential mechanisms contributing to neuronal insult and death in amyotrophic lateral sclerosis.7,16,17
APPROVED MEDICATIONS FOR PEOPLE LIVING WITH AMYOTROPHIC LATERAL SCLEROSIS
Despite substantial investment in research, and a large number of compounds in development, there are currently only four disease-modifying medications approved for ALS, with approval for each being country-dependent.17-24 Three of these medications (riluzole,21 edaravone,22 and PB and TURSO)23 are approved for all forms of ALS. Tofersen, the most recently approved drug, is applicable only to ALS associated with genetic mutations in the SOD1 gene.24,25
Riluzole, approved in the 1990s by several agencies, including the U.S. Food and Drug Administration (FDA) in the USA and the European Medicines Agency (EMA), is hypothesised to inhibit excitotoxicity through anti-glutamatergic properties, and has been shown to modestly improve survival in people with ALS.18,26 Edaravone, approved in a few countries, including Japan and South Korea (2015) and the USA (2017), is hypothesised to suppress oxidative stress, and was shown to slow down disease progression in a cohort of people with ALS with specific disease characteristics.18,27,28 PB and TURSO is a fixed-dose formulation of sodium phenylbutyrate (PB) and ursodoxicoltaurine, and is known in the USA as taurursodiol (TURSO) or by the chemical name tauroursodeoxycholic acid. It was fully approved for treating ALS in the USA, and was approved with conditions in Canada.18 This combination will be the focus of the remainder of the article. More recently, in 2023, the antisense oligonucleotide tofersen received accelerated approval in the USA for adults with SOD1-associated ALS. Accelerated approval was based on reduction in neurofilament levels, a marker of neurodegeneration.24,29
Sodium Phenylbutyrate and Ursodoxicoltaurine
Preclinical and early clinical experience with monotherapy studies
Among the pathogenic mechanisms thought to underlie ALS (Figure 1), motor neuron degeneration may be associated with altered communication between the mitochondria and the ER, due to mitochondrial dysfunction and ER stress.7,30 Both ER and mitochondrial accumulation of misfolded proteins can activate unfolded protein responses mediated by stress responsive kinases, and the translation of transcription factors, such as activating transcription factors 4 and 6.31 Preclinical evidence suggests that some of these mechanisms may be modulated by PB and TURSO, as shown in primary skin fibroblasts from patients with ALS.32
PB is a histone deacetylase inhibitor, with actions postulated to include stabilisation of protein folding and reduction of ER stress.33,34 In a mouse model of ALS, PB administration improved motor symptoms and extended survival, due to actions on genes associated with neuronal apoptosis and protein aggregation.35 Using this mouse model, survival was shown in another study to be increased by 12.8% with PB, 7.5% with riluzole, and 21.5% with a combination of these drugs. Grip strength and weight also improved, and there were positive neuropathological changes, including decreased lumbar ventral horn neuronal cell death.36 Notably, a 20-week, multi-site, Phase II, open-label clinical trial of PB in people living with ALS (N=40) found that the compound inhibited histone deacetylation by approximately 50%, and that it was generally well-tolerated.37
Mitochondrial dysfunction, which can be triggered by accumulation of misfolded proteins, such as TDP-43 or superoxide dismutase, can lead to oxidative stress, bioenergetic failure, alterations of calcium homeostasis,38 activation of intrinsic apoptotic pathways,39 and the interferon-mediated innate immune response.40 Therefore, alleviating mitochondrial dysfunction and ER stress is a rational target for drug therapy in ALS.7,16
TURSO has postulated effects on mitochondrial membrane stability, energy production, and prevention of mitochondrial apoptotic pathway activation.41 Interestingly, a 54-week, Phase II, double-blind, placebo-controlled, randomised controlled trial (RCT) of TURSO in 34 people living with early-stage ALS, and also taking riluzole, found significant slowing in the rate of functional deterioration compared with placebo in the TURSO group. TURSO was generally well-tolerated.42 Of note, a Phase III trial testing TURSO is ongoing in Europe, and is expected to offer more insights into the efficacy of TURSO when offered as monotherapy.43,44
Preclinical combination studies
As discussed above, multiple mechanisms are thought to converge to cause motor neuron death in ALS, including combined effects of mitochondrial damage and ER stress. Accordingly, any monotherapy might be insufficient to substantially affect the pathophysiological cascade. Based on this hypothesis, the combination of PB and TURSO (AMX0035) was developed to target two key pathways, and was investigated in pre-clinical studies.32,45,46
In an in vitro study of primary skin fibroblasts from people with sporadic ALS or healthy controls, PB and TURSO administration synergistically, but not individually, led to upregulation of the expression of genes associated with survival during ER stress, increased expression of mitochondrial respiratory chain subunits, and downregulation of metabolites associated with ALS pathophysiology. Differences were also shown between the ALS and healthy control samples, whereby the PB and TURSO combination abolished the transcriptional profiles associated with ALS.32
The CENTAUR trial was a Phase II, randomised, double-blind trial carried out at 25 USA-based centres of the Northeast ALS Consortium (NEALS) from June 2017–September 2019.47 The initial 24 week RCT phase was followed by an OLE phase of up to 42 months from randomisation (Figure 2), which concluded in March 2021.47-49
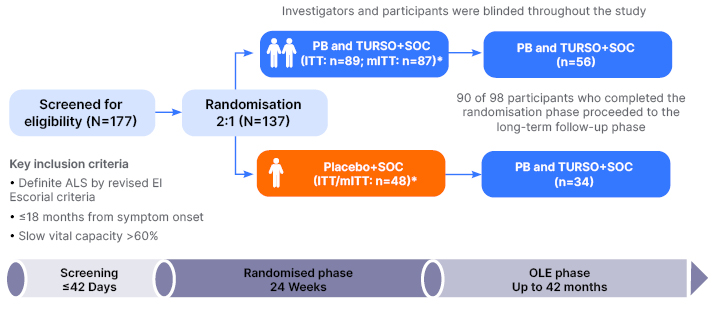
Figure 2: Trial design for the CENTAUR study.47-49
*The mITT population included all participants who received ≥1 dose of PB and TURSO or placebo and had at least one ALSFRS-R total score recorded after randomisation.
ALS: amyotrophic lateral sclerosis; ALSFRS–R: Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; OLE: open-label extension; PB: phenylbutyrate; SOC: permitted standard of care; TURSO: ursodoxicoltaurine/taurursodiol.
PB and TURSO was administered from a pre-mixed, single use powder package dissolved in water, and taken orally or via a feeding tube. During the RCT phase, study drug dosage was once a day for the first 3 weeks, then twice a day. The placebo was matched to PB and TURSO for taste, appearance, and solubility. For the OLE phase, participants received PB and TURSO twice a day with no titration period. In this phase, OLE participants remained blinded to their original RCT treatment assignment. All CENTAUR participants could receive permitted standard of care, which was one or both of the agents approved at the time in the USA for ALS treatment: edaravone and riluzole.47-49
The primary efficacy outcome of CENTAUR was the rate (slope) of decline of the total score on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R) from baseline to Week 24. Secondary outcomes included isometric muscle strength determined by use of an Accurate Test of Limb Isometric Strength (ATLIS) device; plasma levels of phosphorylated axonal neurofilament H subunit, a marker of axonal degeneration;50 slow vital capacity, a measure of respiratory function; time to death, tracheostomy, or permanent assisted ventilation (defined as using ventilation >22 hours daily for >7 days) as individual or composite outcomes; and time to hospitalisation.51 Safety was assessed at each trial visit by assessing any adverse events occurring during the treatment phases.
Baseline characteristics of the randomised participants were similar between the groups (Table 1), except for a slightly lower proportion of edaravone or riluzole use, and slightly higher rates of bulbar onset in the PB and TURSO group compared with the placebo group.47
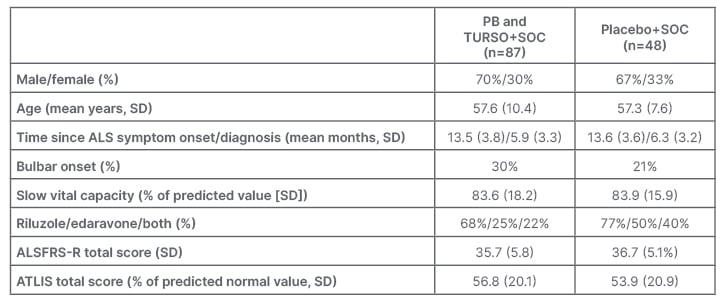
Table 1: Baseline characteristics of the randomised participants (modified intention-to-treat population).47
ALS: amyotrophic lateral sclerosis; ALSFRS-R: Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ATLIS: Accurate Test of Limb Isometric Strength; PB: phenylbutyrate; SD: standard deviation; SOC: permitted standard of care; TURSO: ursodoxicoltaurine/taurursodiol.
A total of 77% of participants in the PB and TURSO group, and 79% of those in the placebo group completed the RCT phase. The primary analysis of functional data was performed in a pre-specified modified intention to treat (ITT) group (n=135), which excluded two participants in the PB and TURSO group who died prior to any postbaseline efficacy assessment. Post hoc ITT analysis (n=137) yielded identical results to modified ITT analysis within rounding error. Survival analyses was carried out in the ITT population.47,48
The study met the primary endpoint at Week 24 (Figure 3). The rate of decline in ALSFRS–R total score was significantly slower in the PB and TURSO group compared with the placebo group, resulting in a 2.32-point absolute difference in ALSFRS–R total score at 24 weeks (p=0.034). Primary analysis results remained consistent after sensitivity analyses that accounted for use of edaravone and/or riluzole. The slope difference between the active and placebo groups was 0.42 points per month (95% confidence intervals [CI]: 0.03−0.81). Thus, treatment with PB and TURSO resulted in an approximately 25% reduction in the slope of functional decline in people living with ALS.47 While there is no consensus on what constitutes a clinically meaningful change in ALSFRS-slope, 25% slowing exceeds the 20% threshold that has been previously identified as clinically meaningful in a survey of 42 ALS researchers and clinicians from the NEALS consortium.52 A previous study showed positive effects of TURSO, when given as monotherapy, on functional outcomes.42 Substantial methodological differences prevent a head-to-head comparison between the two trials, including different sample size, eligibility criteria, duration of follow-up, use of background ALS therapies, and statistical models.42,47
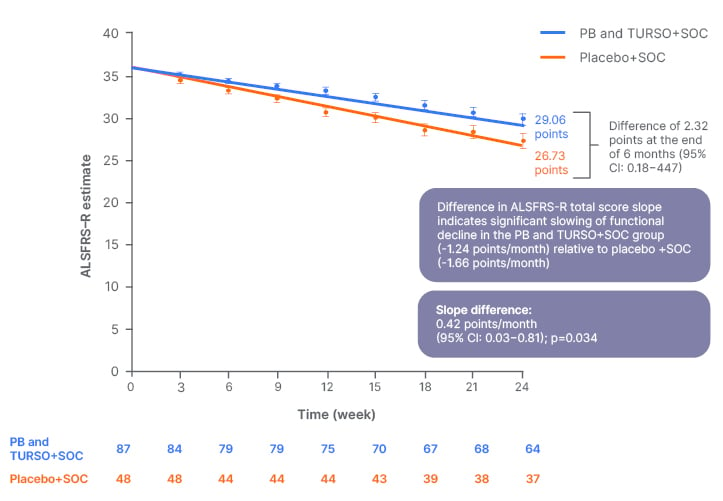
Figure 3: Estimated rate of decline in Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised total score over 24 weeks.47
Adapted from supplemental data from Paganoni et al.47
ALSFRS-R: Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CI: confidence interval; PB: sodium phenylbutyrate; SOC: standard of care; TURSO: ursodoxicoltaurine/taurursodiol.
In terms of muscle strength and respiratory function over 24 weeks, there were no statistically significant differences between the active and placebo groups. Similarly, no significant difference was shown for plasma phosphorylated axonal neurofilament H subunit concentration during the RCT.47 A post hoc analysis of the neuroinflammatory markers chitinase-3-like protein 153 and C-reactive protein showed that their levels were significantly reduced in CENTAUR participants receiving PB and TURSO, compared with placebo and correlated with disease progression.54
Overall, in CENTAUR, the combination of PB and TURSO was safe and well tolerated. Gastrointestinal adverse events, such as nausea, abdominal pain, and diarrhoea, were reported more frequently in the active drug group than in the placebo group, especially during the first 3 weeks of treatment.47 Dose reduction and dose interruption due to gastrointestinal events occurred more frequently in the active group (3% and 9%, respectively) than in the placebo group (0% and 2%, respectively). Five of the PB and TURSO group participants (6%) and two of the placebo group participants (4%) died during the RCT phase.47
Long-term survival and time to event analyses
Survival analyses, conducted up to 42 months after randomisation, included all participants in CENTAUR. They compared participants originally randomised to PB and TURSO to those originally randomised to placebo.48,49 At CENTAUR study end, median survival duration was 4.8 months longer in those originally randomised to PB and TURSO compared to placebo (hazard ratio: 0.64; 95% CI: 0.42−1.00; p=0.048). These results were not impacted by use of riluzole and/or edaravone at baseline.23,49
There was also a significant delay in key ALS progression events in those originally randomised to PB and TURSO, compared with those originally randomised to the placebo group, including longer time to: first hospitalisation (median 17.7 months longer; 40% lower risk); death or death equivalent (tracheostomy/permanent assisted ventilation; median 5.6 months longer; 40% lower risk); all-cause death (median 4.8 months longer; 38% lower risk); or a composite of time to first hospitalisation, death, or death equivalent (4.8 months longer; 38% lower risk).55,56
Post hoc survival analyses evaluating the impact of placebo group crossover in the open-label extension phase
As noted, the above-described survival and time to event analyses compared participants as originally randomised, without accounting for placebo participant crossover in the OLE phase. Indeed, most CENTAUR RCT completers rolled over to the OLE phase (Figure 2), potentially diluting the long-term effects of the drug.49
To evaluate the impact of placebo crossover on the overall survival analysis results, methods were derived from cancer literature, where RCT plus OLE designs are frequently employed. A rank-preserving structural failure time model was applied. This method is designed to estimate what the survival outcome would have been for the placebo group had participants not switched to active treatment.57 The model included the entire randomised population and investigated an endpoint of all-cause mortality at ≤42 months. Using this model, median survival duration difference was 9.7 months (hazard ratio: 0.42; 95% CI: 0.18−0.99; p=0.0048) and the PB and TURSO group had a 58% lower risk of dying versus rank-preserving structural failure time model-adjusted survival in the placebo group (Figure 4).49
Figure 4: Rank-preserving structural failure time model-adjusted survival probability in the sodium phenylbutyrate and ursodoxicoltaurine plus standard of care versus placebo plus standard of care groups.49
Adapted from supplemental data from Paganoni et al.49
CI: confidence interval; HR: hazard ratio; ITT: intention-to-treat; PB: sodium phenylbutyrate; RPSFTM: rank-preserving structural failure time model; SOC: standard of care; TURSO: ursodoxicoltaurine/taurursodiol.
These results are aligned with those of a separate post hoc analysis that leveraged a propensity score-matched external control group from PRO-ACT. The results of this analysis showed a 10.4-month longer median survival duration in PB and TURSO randomised participants compared with the external control group, suggesting a potentially greater survival benefit with PB and TURSO in ALS than seen on the survival analysis that did not consider crossover in OLE.49,58
The Phoenix Phase III Trial of Sodium Phenylbutyrate and Ursodoxicoltaurine
The combination of PB and TURSO was fully approved for the treatment of ALS in the USA, and approved with conditions in Canada in 2022 following findings from the CENTAUR study. To meet regulatory requirements in other regions, and to continue to learn about the effects of the drug on ALS, PHOENIX , a Phase III, double-blind, placebo controlled, multicentre RCT evaluating safety and efficacy of PB and TURSO versus placebo over 48 weeks, was launched.59 The trial enrolled 664 participants from the USA and Europe, and is expected to post results in 2024.59,60 The primary outcomes will include measures of function and survival, as well as measures of quality of life and caregiver burden. Additionally, PHOENIX enrolled a broader population than CENTAUR to allow for more extensive subgroup analyses.59
CONCLUSION
The Phase II CENTAUR trial demonstrated that the combination of PB and TURSO, also known as AMX0035, has beneficial effects in people living with ALS, including slower disease progression and prolonged survival. These effects were seen on top of standard of care medications, and led to the approval of PB and TURSO for the treatment of ALS, thus changing the standard of care for this patient population in selected regions in 2022. The global Phase III PHOENIX trial is expected to add to this body of knowledge by measuring the impact of PB and TURSO treatment in a broader population, over a longer period of time, and on additional outcome measures. PHOENIX trial results are expected in 2024.






