BACKGROUND AND AIMS
The identification of pathological elements that drive disease progression, so-called regulators, is essential when studying progressive diseases. Several neurodegenerative diseases are associated with such disease regulators, key examples being huntingtin in Huntington’s disease and Aβ and τ in Alzheimer’s disease (AD).1,2
When AD presents with symptoms, irreversible neuronal damage has already occurred.3 This fact highlights the need to discover new disease regulators, which can be used for therapeutical development. Targetable intervention and therapy are needed to address presymptomatic processes, and regulatory proteins are a key part of disease progression and represent potential targets.4 Due to the multifactorial pathology of AD, a multimodal approach is needed in order to identify disease regulators. In these studies, the authors used unbiased proteomics coupled with bioinformatics and microscopy methods in order to discover novel disease regulators in AD.
MATERIALS AND METHODS
Proteomics and subsequent Ingenuity Pathway Analysis (IPA) were performed on a knock-in mouse model for AD (AppNL-F/NL-F), which specifically produced increased levels of the neurotoxic peptide Aβ42, making it suitable for studies of Aβ42-driven pathology.
The levels of huntingtin in human and mouse brains were quantified using immunohistochemistry (IHC) and the subcellular huntingtin location was determined by fluorescence microscopy. Potential colocalisation between reactive astrocytes, τ tangles, and huntingtin was determined in human AD tissue by using confocal microscopy.
RESULTS
By using IPA, huntingtin was identified as a presymptomatic regulator of disease progression in a proteomics data set from cortex and hippocampus taken at four different ages (3, 6, 9, and 12 months) from the AppNL-F/NL-Fmouse. Several known Aβ42-associated proteins were also detected. By using semi-quantitative IHC, huntingtin levels were shown to be highly increased in AppNL-F/NL-Fcompared with wild type mice at 6 months of age (3 months before the appearance of amyloid plaques). The huntingtin levels increased over time in wild type mice but not in AppNL-F/NL-Fmice and at 25 months, there was no difference in the levels of huntingtin. Moreover, IHC showed increased levels of huntingtin in pyramidal neurons in the hippocampus and frontal cortex in patients with AD relative to controls (Figure 1).
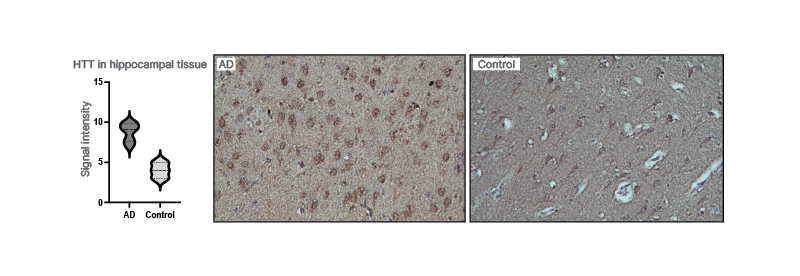
Figure 1: Huntingtin in the brain of patients with Alzheimer’s disease.
Huntingtin is increased in the hippocampal tissue when compared with controls.
AD: Alzheimer’s disease.
In contrast to Huntington’s disease, huntingtin did not colocalise with reactive astrocytes in AD brain, as demonstrated by confocal microscopy. Huntingtin levels were increased in pyramidal neurons in AD brain. There was no significant colocalisation of huntingtin with phosphorylated τ.
CONCLUSION
Huntingtin accumulates in the brain of patients with AD and is distinct from the huntingtin accumulation seen in Huntington’s disease. Huntingtin accumulation precedes AD pathology in AppNL-F/NL-Fmice.
These findings implicate huntingtin in the pathology of AD and warrant further studies. Huntingtin regulation could be a potential treatment for AD due to the numerous clinical trials aimed at Huntington’s disease. The process by which huntingtin regulates AD pathology needs to be established. Mechanistic studies of huntingtin in neuronal cell cultures will shed light on the relationship between huntingtin and AD pathogenesis.