Meeting Summary
Fabry disease, also known as Anderson–Fabry disease, is a rare, inherited X-linked lysosomal storage disorder caused by deficient activity of α-galactosidase A, due to mutations in the GLA gene. Diagnosis can pose a challenge, particularly in females and patients with non-classic or late-onset Fabry disease, given the phenotypic heterogeneity and lack of awareness among healthcare providers, resulting in delayed diagnosis.
This article provides a review of sessions that took place at the Global Fabry Academy 2023, held in-person in Berlin, Germany, in March 2023. The aim of the Academy, a Sanofi (Paris, France)-funded medical educational event, was to provide insights for healthcare professionals, especially nephrologists and cardiologists, to improve the diagnosis and lives of those living with Fabry disease.
During the session ‘Beyond therapeutic goals’, Alberto Ortiz, Professor and Chief of Nephrology and Hypertension at the Research Institute of the Jiménez Díaz Foundation, Madrid, Spain, proposed an individualised approach to the holistic management of both male and female patients with classic Fabry disease to ensure definition of appropriate treatment goals and optimisation of long-term management. Two concurrent sessions focused on the diagnosis of Fabry disease centred on cardiology and nephrology perspectives. Sandra Marques e Silva, an echocardiologist at the Instituto Hospital de Base do Distrito Federal, Brasilia, Brazil, discussed the importance of genetic testing with next-generation sequencing (NGS) from the cardiology perspective in the diagnosis of hypertrophic cardiomyopathy (HCM) and Fabry disease. Maurizio Pieroni, a cardiologist at the San Donato Hospital Arezzo, Italy, discussed the importance of cardiac imaging, and reviewed the importance of left ventricular mass index to guide Fabry disease diagnosis in patients with unexplained HCM. Liffert Vogt, Professor of Clinical Nephrology and Renal Physiology at the University of Amsterdam, the Netherlands, discussed the changing prevalence and unknown causes of end-stage kidney disease (ESKD), proposing a role for NGS in chronic kidney disease (CKD) in the context of Fabry disease, while Antonio Pisani, Nephrologist at the University Federico II of Naples, Italy, considered the significance of parapelvic cysts in the diagnosis of Fabry disease.
The plenary session, co-chaired by Christine Kurschat, Professor of Medicine at the University of Cologne, Germany, and Christoph Wanner, Professor of Medicine and Head of the Division of Nephrology at the University Hospital of Würzburg, Germany, featured a discussion with patient representative Natascha Sippel-Schönborn who provided a patient’s perspective on Fabry disease, while the physicians’ perspective presented by Wanner served as a comparison. The conversation brought to light the challenges of diagnosing Fabry disease, particularly in females, as well as the disease’s impact on patients’ quality of life (QoL), and the need for holistic care and psychological support. The session highlighted the importance of a patient-centred approach to care for individuals with Fabry disease.
Beyond Therapeutic Goals in Patients with Classic Fabry Disease
Alberto Ortiz
Fabry disease can present as classic or non-classic forms. Classic Fabry disease is well defined in males and is associated with symptoms such as neuropathic pain, gastrointestinal manifestations, hypohidrosis, angiokeratoma, and persistent fever starting in childhood.1-3 In older male children, albuminuria develops (signalling the onset of CKD), progressing to proteinuria and loss of kidney function, ultimately leading to kidney failure. Cardiac disease also develops, and cerebrovascular disease may occur.1,2 Kidney events usually precede cardiac events in males, and a correct understanding of this sequence of events facilitates the interpretation of clinical trial data (as described below). Thus, the median age of kidney failure is similar for males and females (40 years), but kidney failure is approximately 20-fold more common in males.1
Ortiz identified that the symptoms of classic Fabry disease in females are highly variable due to being “mosaics of Fabry and healthy cells,” which may result in some individuals having more Fabry cells than others.4 Ortiz stated this variability makes it difficult to identify females with Fabry disease who require therapy, and also to assess the efficacy of therapy in clinical trials.4,5 However, at the individual Fabry cell level, females are as severely affected as males, with the same amount of glycolipids, and, according to Ortiz, should receive the same intensity of therapy as intended for males with the same genetic variant.4,5
The European Fabry Working Group has established consensus recommendations for assessing and treating Fabry disease.6 Diagnosis should be followed by a comprehensive initial patient assessment to evaluate organ pathology, as well as signs and symptoms such as renal (microalbuminuria, proteinuria, and estimated glomerular filtration rate), cardiac (left ventricular hypertrophy [LVH], fibrosis, and 24-hour cardiac rhythm disturbances), central nervous system (white matter lesion, clinical evidence, hearing loss), neuropathic pain, and gastrointestinal involvement.6 The identification of any one of these should prompt therapy initiation. Ortiz emphasised that individualised therapeutic goals should be established, and personalised therapeutic management based on those goals, which should be discussed with the patient, family, and carers (for paediatric patients).6
Enzyme replacement therapy (ERT) is the standard treatment for classic Fabry disease. There are two preparations of ERT available in most countries: agalsidase-α (0.2 mg/kg every other week) and agalsidase-β (1 mg/kg every other week), which achieve differing intracellular enzymatic activity.7 Ortiz discussed evidence from the Canadian Fabry Disease Initiative (CFDI) comparing these two doses, which demonstrated no difference in cardiac or neurological events or mortality, stating that the study recruited a small fraction of the initially planned patients, therefore conclusions cannot be reliably drawn.8 Furthermore, there was no difference in renal events or kidney function in females, but these are uncommon in female patients with unselected Fabry disease, even among those who have disease manifestations severe enough to initiate ERT.8 A key result of the trial was a differential impact of the agalasidases on the most frequent events, which were the kidney events (nephrotic range proteinuria, doubling of serum creatinine, or kidney failure) in males. Indeed, the incidence of kidney events in males randomised to receive agalsidase-β was 0.31 events per 100 patient months, whereas those receiving agalsidase-α was 1.1 events per 100 patient months.8 This resulted in an incidence rate ratio of 0.24 (p=0.006).8 Ortiz indicated that differences in kidney outcomes should be considered when deciding on the specific Fabry therapy.
The recommendations for identifying patients with classic Fabry disease include genetic screening and assessment of glycolipid burden through measuring globotriaosylsphingosine (lyso-GL-3).6 Regarding treatment outcome assessment, Ortiz suggested moving away from repeat biopsy and using lyso-GL-3 (routine monitorisation at baseline, then every 6 months) after treatment initiation or switch to evaluate longitudinal changes. It is also important to monitor neutralising antibodies alongside lyso-GL-3 levels in patients receiving ERT. The highest antibodies against ERT are typically seen in male patients with classic Fabry disease and truncating genetic variants, i.e., in persons who lack endogenous α-galactosidase A. In an open-label, multicentre, exploratory, Phase IV study, switching from agalsidase-α to -β resulted in a mean percentage reduction of 40% in lyso-GL-3 from baseline (p<0.001) after a period of 6 months (clinical outcomes were not assessed in this study).9
For cardiac response, ECG, echocardiogram, cardiac MRI, and biomarkers should be assessed, with improvements in most measures except fibrosis, which should not worsen. For renal function, albuminuria should be assessed in mg/g or mg/24h, a urinary albumin:creatinine ratio >30 mg/g is considered ‘the magic number’ to define kidney disease according to Ortiz, and the target should be to lower albuminuria below 30 mg/g. Estimated glomerular filtration rate should also be assessed, whereas proteinuria is not a good early marker due to urinary dilution, as urine concentration defects are an early feature of Fabry disease.10 Additionally, cerebrovascular MRI should be performed.6
Treatment and clinical status should be regularly monitored, using baseline data for comparison, every 6 months to assess whether therapeutic goals are being achieved.6 Once therapeutic goals for patients with classic Fabry disease have been achieved, the next step is maintaining disease-specific and concomitant treatments.6 Ortiz recommended that if therapy is ineffective, specific and non-specific therapy may need to be adjusted, including the consideration of switching therapy to achieve therapeutic goals.6
Ortiz summarised that, once the initial targets have been achieved for patients with classic Fabry disease, the focus can shift to addressing remaining pathological gaps through an individualised approach to the holistic long-term management.
Ortiz concluded that although the phenotype of Fabry disease may vary, the severity of Fabry cell involvement is the same in both males and females. Therefore, males and females with the same genetic variant should receive the same intensity of therapy. The earliest objective response to therapy is typically a decrease in circulating lyso-GL-3 within a few months. If individualised therapeutic goals are not met, it should prompt an early reassessment of the therapeutic approach to ensure the best possible outcome for the patient.
Fabry Disease Diagnosis in Cardiology
Next-Generation Sequencing
in Cardiology
Sandra Marques e Silva
In HCM, genetic testing has been shown to have a beneficial impact on the diagnosis and clinical management, and is a Class I recommendation in clinical guidelines.11,12 Marques noted that genetic testing plays a crucial role in identifying HCM phenocopies and differential diagnoses, such as Fabry disease, and family screening and cascade testing of family members are crucial to identify individuals requiring treatment.12,13 Marques highlighted that guidelines indicate that genetic testing is recommended in patients fulfilling diagnostic criteria for HCM to enable cascade genetic screening of their relatives. If red flags are apparent based on clinical manifestations, ECG, cardiac imaging, or laboratory findings (Figure 1), Fabry disease should be considered, and clinicians should direct patients to genetic testing.13 Marques mentioned that genetic counselling before and after testing is recommended.
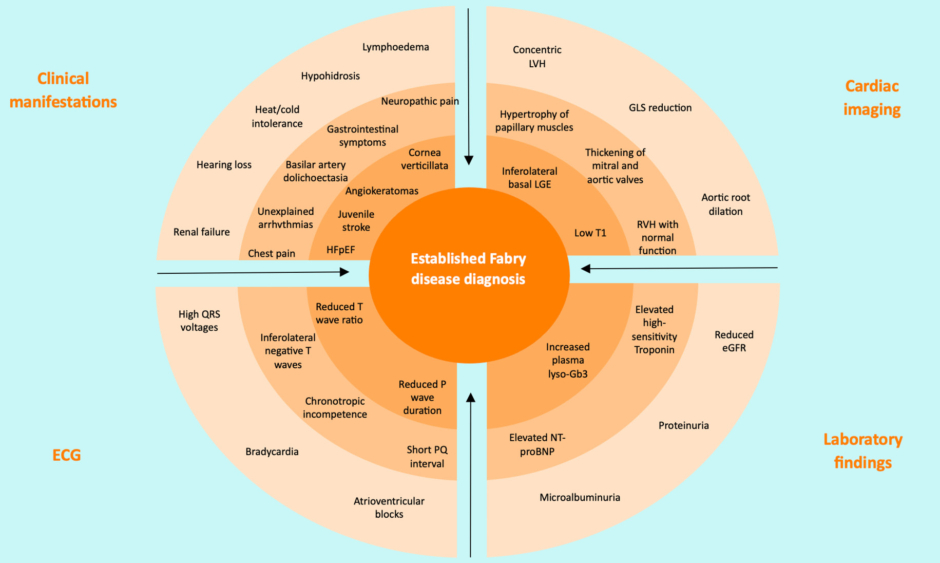
Figure 1: Red flags in establishing Fabry disease diagnosis.13
Increasing likelihood of Fabry disease diagnosis from outer and/or brighter to inner and/or darker circles.
eGFR: estimate glomerular filtration rate; GLS: global longitudinal strain; HFpEF: heart failure with preserved ejection fraction; LGE: late gadolinium enhancement; LVH: left ventricular hypertrophy; lyso-Gb3: globotriaosylsphingosine; NT-proBNP: N-terminal pro-brain natriuretic peptide; RVH: right ventricular hypertrophy.
Adapted from Pieroni et al.13
Marques identified that genotyping is particularly necessary for the diagnosis of Fabry disease in female patients, as α-galactosidase A enzyme activity does not define the disease.13 Genotyping can help predict phenotypic expression (classic or late-onset), as well as identifying at-risk and asymptomatic carriers, which Marques noted helps expedite treatment.14,15 As well as aiding decision-making for treatment of the individual, genetic testing enables pre-conception genetic counselling.15 Marques noted that genotyping is necessary to receive treatment in Brazil.
Marques presented four case examples (Table 1) that demonstrate the benefits of NGS panel testing in cardiology. All patients presented with similar symptoms of dyspnoea and thoracic pain on exertion, only differing by sex, age, and family history. These cases were originally identified with HCM and LVH. Various diagnostic tests were used, including ECG, echocardiogram, MRI, and genotyping. The proposed diagnoses included Fabry disease, HCM, or other conditions such as Chagas disease or amyloidosis.
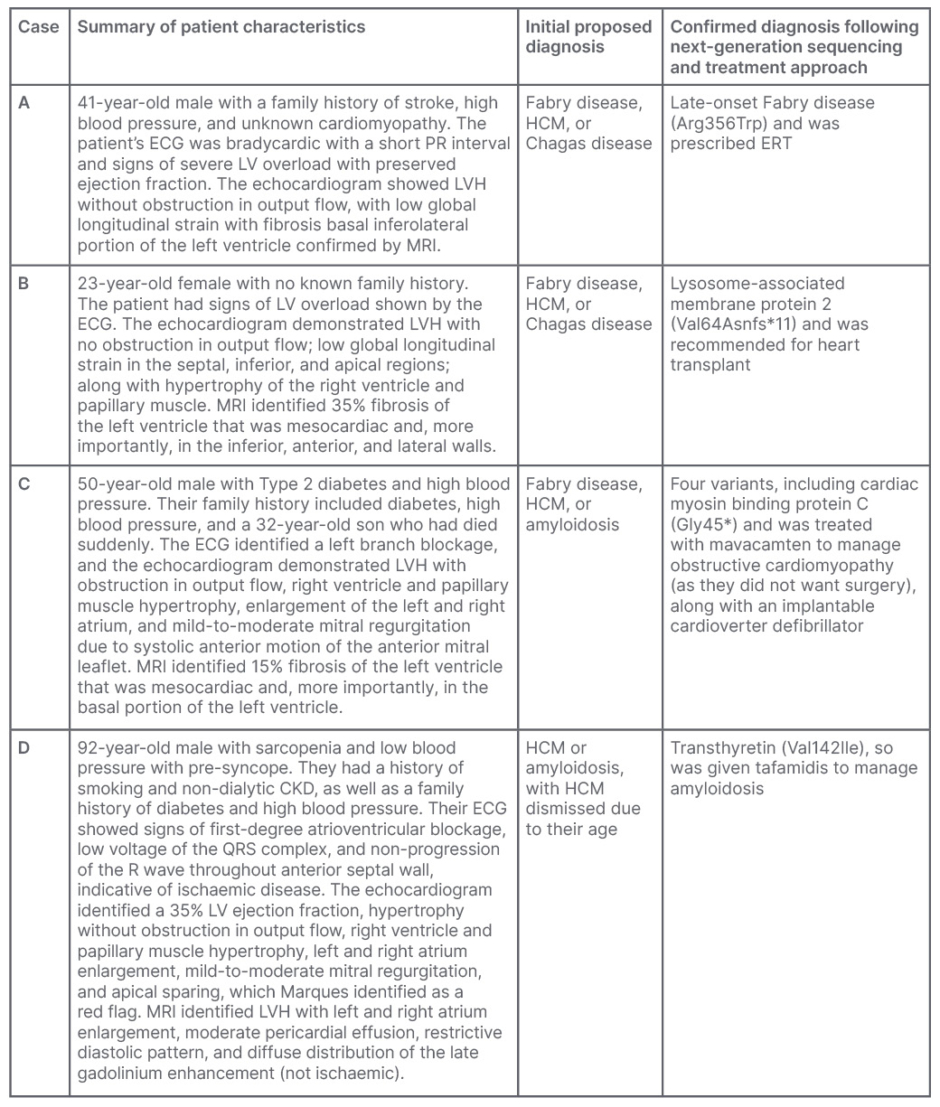
Table 1: Summary of clinical cases that underwent cardiological assessment and next-generation
sequencing to determine diagnosis.
CKD: chronic kidney disease; ERT: enzyme replacement therapy; HCM: hypertrophic cardiomyopathy; LV: left ventricular; LVH: left ventricular hypertrophy.
Medical information provided from Marques e Silva’s patient database with the patients’ authorisation.
Marques noted that without NGS, these patient cases would have received various treatments for symptoms of heart failure, arrhythmia, ischaemia, and embolism risk.16 However, NGS allowed for the identification of specific genetic mutations that enabled physicians to prescribe targeted treatments. The diversity of treatments employed following NGS included ERT for late-onset Fabry disease, heart transplant for Danon disease, mavacamten to manage obstructive cardiomyopathy, and tafamidis to manage amyloidosis, highlighting the value of differential diagnoses aided by NGS panels.
Marques concluded by summarising the benefits of using NGS panel testing in cardiology for faster and more accurate diagnosis, leading to better treatment and genetic counselling, stating that while physicians try to do their best, “our best can be even better if we have the panel.” Marques also highlighted the importance of HCM and Fabry disease diagnosis, especially in females and clinically silent, genetically affected family members, allowing for informed decisions about patient care.
Role of Left Ventricular Mass Index in Unexplained Hypertrophic Cardiomyopathy
Maurizio Pieroni
Hypertrophic cardiomyopathy is typically characterised by an increased LVM index.17 In patients with HCM older than 35–40 years, the prevalence of Fabry disease is approximately 0.5–1.0%.11 When considering the differential diagnosis of HCM, Pieroni suggests adopting a ‘cardiomyopathy mindset’.18 This approach can help generate suspicion of specific aetiologies and identify red flags (Figure 1).13
According to Pieroni, cardiac imaging using echocardiography is crucial in the initial assessment of HCM with suspected Fabry disease, as a more precise diagnosis can be made.13,19 Advances in cardiac MRI, specifically the use of T1 mapping tools, have changed the approach to identifying HCM phenotypes.20,21 Low T1 values are typically observed in Fabry disease, with most patients falling below the normal T1 threshold, making it important in early diagnosis and screening family members for cardiac involvement in Fabry disease.20,21 Pieroni stated that “T1 is central in the differential diagnosis of patients with unexplained ventricular hypertrophy,” including late-onset Fabry disease or in patients without systematic red flags. Therefore, cardiac MRI with T1 mapping can identify red flags and direct the physician towards genetic testing.
While reducing LVM has historically been the primary target for HCM treatment, Pieroni argued that the pathophysiology of cardiac damage in Fabry disease is more complex and involves other mechanisms triggered by lysosomal storage, including sarcomere, mitochondrial, and ion channel dysfunction, apoptosis, and inflammation.13 As a result, early initiation of ERT through screening and early diagnosis, preventing lysosomal storage at an earlier stage, can also prevent other manifestations of Fabry disease.13 As an example of other disease-modifying treatments in cardiomyopathies, Pieroni presented evidence that myosin inhibitors target the pathophysiology of sarcomeric HCM at a cellular level by reducing haemodynamic biomarkers and troponin levels.22 Pieroni also noted that echocardiography may not be reliable in measuring LVM index, questioning the reliability of the formula used by devices and demonstrating inter-rater variability among technical operators.23 Cardiac MRI may provide a more comprehensive assessment of cardiac effects of treatments allowing a more precise measurement of LVM together with tissue characterisation.
Pieroni concluded that the focus should not be solely on hypertrophy through LVM index for diagnosis, prognosis, and assessment of efficacy of new treatments in Fabry disease. Instead, more complex cardiac endpoints, including clinical endpoints, cardiac and Fabry disease-specific biomarkers, QoL, functional capacity testing, and advanced cardiac imaging should be used.
Fabry Disease Diagnosis in Nephrology
Next-Generation Sequencing in Nephrology
Liffert Vogt
Vogt discussed the potential of identifying undiscovered patients with Fabry disease within high-risk populations with ESKD. Various diagnostic tests are available; however, Vogt mentioned they may not be applied to the right patients, and misdiagnoses and diagnostic delays are common, leading to progressive and irreversible organ damage. 24,25 Screening dialysis patients for Fabry disease may be ineffective. A multicentre, prospective, observational cohort study of male patients conducted in the Netherlands (>18 years at time of dialysis initiation; N=508) screened using enzymatic assay; it only identified one patient with already known Fabry disease, and no new previously undiagnosed patients.26,27
Additionally, the prevalence of ESKD is changing.28 A Dutch study found that the share of primary diagnosis of glomerulonephritis and pyelonephritis declined by 5% between 2002 and 2017, whereas Type 2 diabetes and hypertension increased by 3%, and the proportion of cases with unknown causes of ESKD increased by 2%.28 A kidney biopsy and use of electron microscopy to confirm Fabry disease nephropathy is often required. At high magnification, but preferably at electronic microscopy, intralysosomal inclusion bodies, indicative of classic Fabry disease, can be identified.29 However, Vogt stated that kidney biopsy will not be helpful in most circumstances, mainly because large scarring impairs interpretation, in cases of progressive CKD attributed to diabetes, hypertension, or nephrosclerosis of unknown cause.
A USA-based primary prevention study identified a four-times higher relative risk for ESKD associated with hypertension.30 However, the crude rate, or absolute risk, was found to be “very low” (3.4 per 100,000) according to Vogt.30 Vogt explained that as clinicians are now more focused on treating hypertension, this risk is likely to be lower. Therefore, hypertension is not necessarily a causative factor of ESKD, and Vogt questioned whether hypertensive nephropathy diagnosis is the best approach to patient care. Vogt proposed using NGS to identify whether among patients with CKD of unknown causes a primary kidney disease (e.g., Fabry disease) leading to hypertension may have been missed.
Vogt presented evidence utilising genotyping in kidney transplant patients to help identify inherited causes of ESKD. In one example, five international cohorts of patients with adult-onset ESKD (≥18 years or older; N=5,606) were screened for nephronophthisis-related genes.31 Of these, 26 (0.5%) showed homozygous NPHP1 deletions and only 12% (n=3) were correctly diagnosed phenotypic nephronophthisis, whilst most were another cause (88%), including CKD with an unknown aetiology (n=11).31 Another international study screening 335 patients from 300 families, identified that among 74 families, 25% had a causative mutation in one of 20 known genes associated with steroid-resistant nephrotic syndrome.32 The study revealed novel mutations in known genes (3.7%) and novel causative gene mutations (28.0%).32 Vogt highlighted this information “would be useful upfront” considering the clinical consequences of other diagnoses in therapy resistance.
A study in two USA cohorts, screening 600 genes in ESKD (N=3315), identified genetic diagnostic variants in 307 patients (9.3%), including 66 monogenic disorders.33 Patients with a given diagnostic category for congenital or cystic kidney disease revealed a high proportion of diagnostic variants of the gene. However, the spectrum of clinical diagnoses was more varied for other kidney diagnosis categories.
Vogt described the VARIETY study, an ongoing nationwide prospective cohort study in the Netherlands of patients with ESKD with estimated glomerular filtration rate <60 mL/min/1.73m2 at age <50 years, to determine the diagnostic yield of kidney disease (the percentage of participants with a genetic diagnosis) in clinical practice.34 The study includes patients with CKD without a primary kidney diagnosis or with unclear diagnosis (N=1,000).34 Preliminary data from 340 patients (mean age: 45 years; mean age at diagnosis: 34 years) indicated that approximately one-third had a positive family history of kidney disease (Vogt, unpublished data, 2023). In approximately 40% of cases, a kidney biopsy was performed but did not identify the correct diagnosis. The only predictor of kidney disease was family history, underscoring the significance of documenting family history. The study estimated that ≥70% of patients had a genetic diagnosis, the majority being Alport syndrome and nephronophthisis, and one case of Fabry disease (Vogt, unpublished data, 2023). This highlights that NGS has clinical consequences for the majority of patients (approximately 75% [Vogt, unpublished data, 2023]), impacting pregnancy, pre-conception counselling, and genetic family screening. Vogt concluded NGS can be valuable in CKD diagnosis and has significant clinical implications for patients.
Parapelvic Cysts
Antonio Pisani
Over 84% of patients with Fabry disease have renal impairment, typically becoming apparent in their mid-30s, although it can occur in childhood.35-37 In the absence of a specific treatment, renal failure is a major cause of
Fabry disease mortality, shortening life expectancy of males and females by 20 and 15 years, respectively.36,38
While CKD in Fabry disease accounts for 0.01% of ESKD cases in Europe and the USA, the true prevalence may be higher, according to Pisani.10 In a systematic search of dialysis screening (N=39,621), 0.23% of male (n=91) and 0.06% of female (n=25) patients were positive for the GLA gene.39 GLA variants were three-to-four times higher than expected, at 0.24% (95% confidence interval: 0.17–0.32), with pathogenetic mutations of 0.14% (95% confidence interval: 0.08–0.20).39
Although the pathogenesis of Fabry disease remains complex and not fully understood, the lysosomal accumulation of globotriaosylceramide (GL-3) initiates a cascade of events leading to lysosomal and vascular damage, resulting in oxidative stress, inflammation, and immune dysfunction.,40–44 These lead to fibrosis and renal damage, affecting various renal cell types (podocytes, tubular cells, glomerular endothelial, mesangial, and interstitial cells). 44-47
Pisani proposed that the presence of parapelvic cysts indicates specific renal abnormalities for early diagnosis of Fabry disease, before kidney damage occurs.48 Parapelvic cysts arise within the renal parenchyma adjacent to the renal sinus. They account for 2.8–6.0% of all renal cysts in adults, but do not correlate with renal function or kidney injury, nor increase with age.49,50 Pisani pointed out that parapelvic and peripelvic cysts are completely different, not only due to their localisation, but also for their number, dimensions, and origin. Specifically, parapelvic cysts are usually single, large cysts, whereas peripelvic cysts are more numerous and smaller.49,50
Pisani noted that parapelvic cysts had previously been associated with Fabry disease nephropathy, being more common and appearing earlier than in the general population.51-53 A seminal study of patients with hemizygous Fabry disease (N=24) demonstrated an increased prevalence of parapelvic kidney cysts.54 Pisani highlighted that despite the study limitations, Fabry disease should be considered in the differential diagnosis of patients with multiple renal sinus cysts and kidney disease.54
An Italian multicentre, retrospective study (N=173) explored the prevalence of parapelvic cysts using renal ultrasound in patients with Fabry disease, who were age-, sex-, and renal function-matched with controls.55 The frequency of parapelvic cysts was significantly higher in patients with Fabry disease (n=50 out of 173 [29%]) compared with controls (n=2 out of 173 [1%]; p<0.001).55 No differences in the presence of other renal abnormalities, such as cortical cysts, were found.55 Using a more accurate ultrasound increased parapelvic cyst detection by 14% (20 out of 67 to 29 out of 67). However, 58% (n=93) had no abnormality detected (p>0.05).55 Pisani noted both cortical and parapelvic cysts were present at a young age, with no difference between sexes.55
Pisani recognised that the exact aetiology and mechanisms behind parapelvic cyst formation in Fabry disease remain to be elucidated, and it is yet to be determined whether parapelvic cysts affect storage or vice versa.54,55 Pisani hypothesised an association with glycosphingolipid metabolism.56 Nevertheless, Pisani concluded, a high prevalence of parapelvic cysts in classic Fabry disease suggests, while not considered a pathognomonic sign, their presence should prompt nephrologists and radiologists to consider Fabry disease.
Heterogeneity of Fabry Disease: Managing the Expectations of Physicians and Patients
Christine Kurschat, Christoph Wanner, and Natascha Sippel-Schönborn
The plenary shared valuable insights on the challenges faced by patients with Fabry disease. A patient’s journey towards diagnosis can take on average 14 years from onset of symptoms for males and 16 years for females, resulting in therapy delay.57 Kurschat asked Sippel-Schönborn, who has Fabry disease, about their experience, which highlighted the lengthy diagnosis process that many female patients face. Sippel-Schönborn had initially been diagnosed with a heart attack after experiencing breathing difficulties and elevated troponin values. Ten years prior, Sippel-Schönborn had developed heart problems that were unexplained at the time. Their family history revealed their father’s hypertension and LVH. When Sippel-Schönborn informed their physician of their father’s renal problems and cause of death (heart attack), it raised suspicion about an additional underlying cause of their elevated troponin values. Sippel-Schönborn was diagnosed with Fabry disease at the age of 52 years, which they said “made a change” to them.
Fabry disease not only affects vital organs such as the heart, kidney, and brain, but also impacts QoL.46 Symptoms such as anxiety, depression, pain, and reduced wellbeing, may often be overlooked by physicians. When asked by Kurschat about their experience, Sippel-Schönborn indicated that issues of pain were not taken seriously or were misinterpreted.46,58 Kurschat asked if they had any recommendations for physicians communicating with patients. Sippel-Schönborn responded that physicians should take patients’ complaints seriously, especially in females, when they report symptoms that physicians cannot explain, and consider their overall wellbeing. Sippel-Schönborn emphasised the importance of being understood after a long period of struggling to be diagnosed. Physicians should not dismiss symptoms if unrelated to their expertise and should try to connect the dots. Wanner stated that physicians may not always know what is in front of them but have to be careful and follow standard operating procedures.
Kurschat noted patients often experience anxiety and fear related to their diagnosis, including the burden of integrating treatments such as ERT infusions into their lives. A meta-analysis evaluating the impact of Fabry disease on QoL (N=599), demonstrated a reduction of QoL compared with the general population.58 Kurschat also noted that this was impacted by factors such as the use of chronic medications and the need for recurrent medical appointments. Patients may require psychological support, which is often insufficient and difficult to access in most countries. Kurschat’s opinion was that it is often hard for physicians to give support to improve QoL. Sippel-Schönborn stated that it is important, as a patient, to manage one’s own QoL, but they had observed that experiencing chronic pain had led to depression, anxiety, and further deterioration of QoL in other patients.
The discussion also raised the issue of genetic variants of unknown significance. Some gene mutations identified in the GLA gene have been found to be benign mutations.59 Kurschat noted that for some patients who were initially diagnosed with Fabry disease, genetic testing revealed these polymorphisms. Kurschat stated that this caused uncertainty and fear among those patients who were hesitant to discontinue treatment. Sippel-Schönborn said there was a great deal of insecurity among patients who come to Fabry disease patient association support groups seeking answers and assistance. Sippel-Schönborn emphasised the importance of ongoing research, discussion, and continued support for patients.
Wanner provided the physician’s perspective on Fabry disease, suggesting that diagnosis is often difficult. Wanner said it is only “relatively easy” to diagnose when Fabry disease is suspected with unexplained cardiovascular events in younger individuals.46 Wanner highlighted that physicians struggle with questions about who to treat, when to start treatment, which treatments to use, the role of genetic variants, and what adjunctive therapy and support are available. Furthermore, Wanner noted, many symptoms of pain experienced by patients may not necessarily be due to Fabry disease.46
Wanner highlighted a couple of critical points. Firstly, physicians “do not prolong” the patient’s journey; instead, they refer the patient as soon as they associate the patient’s symptoms with disease. However, physicians often do not know to where they should refer the patient. Secondly, Wanner emphasised the importance of evaluating the patient’s family tree, and that it would be logical to evaluate more family members once an index case is identified.46 However, physicians do not always follow this practice, as it is a cumbersome and lengthy process that is not adequately remunerated. Moreover, Wanner agreed that physicians tend to focus on clinical events, while other symptoms, QoL, or hospital admissions are considered secondary. Patients’ overall wellbeing is often seen as the patient’s responsibility. Wanner mentioned that physicians may use the ‘fire and forget’ concept of prescribing treatment and moving on, but annual disease progression workup is necessary to determine whether more care, adjunctive treatment, or supportive treatment is necessary, as recommended by international guidelines.46 Wanner also noted the importance of registering patients with Fabry disease with a specialist centre that can provide annual care through a multidisciplinary team.59
Prescribing information for Sanofi B.V. products mentioned in this article: Fabrazyme (agalsidase beta) can be found here. Always consult local prescribing information in country of practice as information may vary.
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: http://www.mhra.gov.uk/yellowcard or search for MHRA Yellow Card in the Google Play or Apple App Store. For non-UK HCPs, suspected adverse reactions should be reported via national reporting system in country of practice.
Compliance number: MAT-GLB-2303028-1.0 – 07/2023