Abstract
Previously, membranous nephropathies were divided into primary and secondary categories when the exact mechanism or pathogenetic factor were unknown. Approximately 70% accounted for primary membranous nephropathies. The remaining 30% were called secondary because they developed due to well-known diseases such as autoimmune diseases, tumours, infections, or drug assumptions. The discoveries of the M-type phospholipase A2 receptor and of thrombospondin type 1 domain containing 7A as causative antigens in a part of the so-called primary membranous nephropathies opened new knowledge on the effective causes of a large part of these diseases. The availability of novel techniques such as laser micro-dissection and tandem mass spectrometry, as well as immunochemistry with antibodies directed against novel proteins, allowed the confirmation of new antigens involved. The use of confocal microscopy and Western blot allowed detection of the new antigen on glomerular membrane, and the same antigen and relative antibodies have been detected in serum samples.
Through these techniques, four new antigens were first detected, including neural epidermal growth factor 1 and semaphorin 3B in the so-called primary membranous nephropathy, and exostosin 1 and 2 and neural cell adhesion molecule 1 in lupus membranous nephropathy.
The aim of this study is to describe the characteristics of the new antigens discovered and their association with other diseases. In addition, new antigens are on the horizon, and the story of primary membranous nephropathy is still to be completely written and understood.
Key Points
1. Until 2002, most of the antigens that were involved in membranous nephropathy (MN) were unknown; however, other antigens have been discovered since then.
2. By using novel techniques, the authors were able to identify approximately 1,500–2,000 proteins in glomerular extracts and also four new antigens associated with MN.
3. The histological pattern for all of the primary MNs is similar, despite being characterised by different clinical and outcome findings.
INTRODUCTION
Membranous nephropathy (MN) is an autoimmune disease in which autoantibodies directed against antigens localised on the glomerular basement membrane (GBM) activate the complement cascade and cause severe proteinuria.
The histology of the disease is similar, but the causes of MN may be quite different. Indeed, the disease may occur as primary MN without the concomitance of other associated disease (80% of cases); or secondary MN when it is associated with different diseases, such as infections, autoimmune diseases, and cancers.1,2 Until 2002, the vast majority of the antigens involved in the primary MN were unknown. The aim of this review is to highlight the recent pathophysiological advances that allowed the identification of many of the antigens involved.
FIRST ANTIGENS DISCOVERED
The first antigen discovered as cause of MN was the neutral endopeptidase (NEP). This is an infrequent cause of MN in newborns.3 The disease developed because females who were pregnant had a genetic abnormality leading to NEP deficiency, caused by a mutation in the MMF gene that codes for NEP.4 This fact causes alloimmunisation during pregnancy, and transplacental antibodies passing to the fetus, principally by the end of pregnancy.
More recently, in 2009, researchers found that many patients affected by primary MN have antibodies in the glomerular eluate against an epitope of the phospholipase A2 type M receptor (PLA2R), documenting that this protein is an important cause of MN.5 Almost simultaneously, other researchers6 found a colocalisation of antibodies against anti-aldose reductase and anti-manganese superoxide dismutase (SOD2) with IgG4 and C5b-9. These data suggest that anti-aldose reductase and SOD2 are antigens responsible for MN, and that the oxidative stress may induce the SOD2 expression in the glomeruli.
Endopeptidase neutral and PLA2R are antigens that, normally, are localised on podocytes. In adults, high levels of antibodies anti-NEP are not a sign of pathology. On the contrary, high levels of antibodies against PLA2R are sign of pathology, and help in differentiating primary MN from secondary MN.7
On 2014, Tomas et al.8 found that up to 5% of patients affected by primary MN have antibodies against thrombospondin-type 1 domain containing 7A (THSD7A). THSD7A is localised on the basal portion of podocytes, and colocalises with nephrin.9 Although PLA2R and THSD7A were considered to characterise primary MN, later PLA2R-related MN was documented to be also associated with hepatitis B infection10 and sarcoidosis.11 Additionally, THSD7A-related MN may be associated with cancer and the antigen has been found in tumour cells.12 PLA2R and THSD7A were involved in 7% and 5% of primary MN (Figure 1).
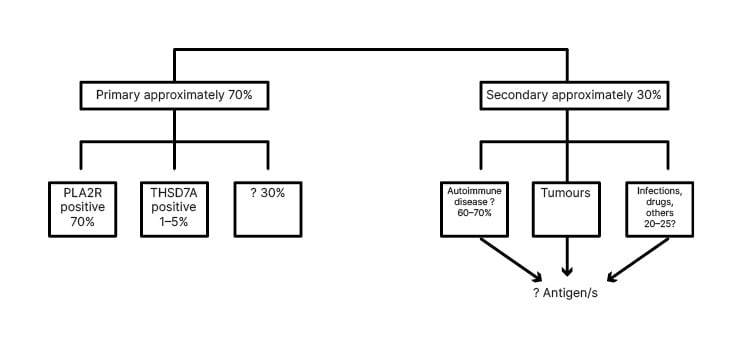
Figure 1: Older classification of membranous nephropathies.
PLA2R: phospholipase A2 type M receptor; THSD7A: thrombospondin-type 1 domain containing 7A.
NOVEL TECHNIQUES
Thereafter, laser micro-dissection of the glomeruli, followed by tandem mass spectrometry (MS/MS) was introduced as a new technique by Sethi et al.13 In this way, novel antigens examining trypsin-digested proteins could be identified. With the use of this technique, the authors could also use paraffin-embedded biopsies, allowing the identification of the antigen even in patients with immunologically inactive diseases. These novel techniques allowed the identification of approximately 1,500–2,000 proteins in glomerular extracts, and allowed a semi-quantitative measurement.
The study started from PLA2R negative biopsies. The identification of a new antigen in these trypsin digested micro-dissected glomeruli was carried out with antigen detection in subepithelial space by immunohistochemistry. MS confirmed the nature of the antigen in the biopsies. To confirm the new findings, European cohorts validated the data. Due to these techniques, four additional antigens were found between 2019 and 2020: neural cell adhesion molecule 1 (NCAM-1), exostosins 1 and 2 (EXT1/EXT2), neural epidermal growth factor 1 (NELL-1), and semaphorin 3B (SEMA 3B).13,14-16 New antigens will be discovered in the future.17 Figure 2 shows the molecular structures of these antigens compared with PLA2R and THSD7A. While the molecular structure will be described in chapters concerning the different antigens, Figure 2 is useful in understanding which antigens are more typical of primary MN, and their capacity to activate the complement pathways.
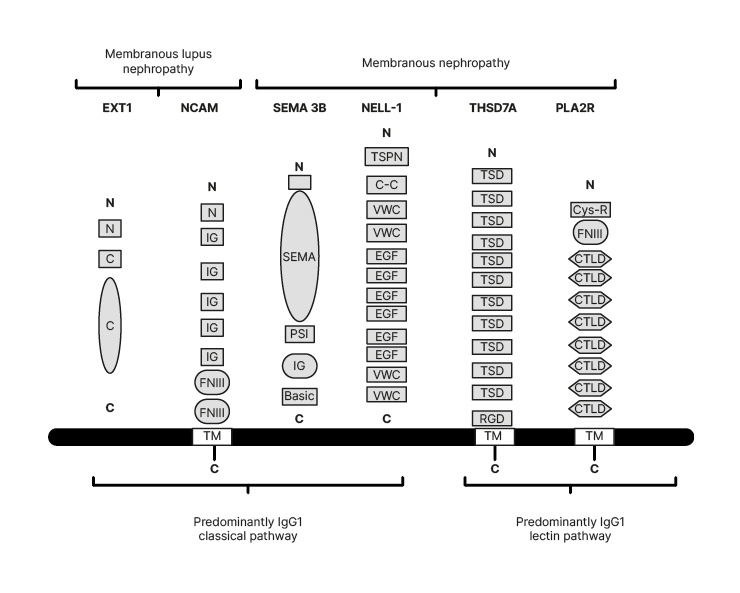
Figure 2: The molecular structures of the four new antigens compared with the old antigens.
Exostosins (EXT1) are transmembrane proteins that have a short amino-terminal cytoplasmic tail, a single transmembrane domain, and a long globular catalytic C-terminal domain. NCAM-1 contains an aminoterminal tail, five IgG domains, two FNIII domains, and a TM domain. SEMA 3B has large SEMA domain region, a PSI domain, an Ig domain, and a short C terminal basic domain.
C: C-terminal domain; C-C: coiled-coil domain; Cys-R: cys-rich domain; CTLD: C-type ltctin-like domain; EGF: epidermal growth factor; EXT1: exostosin 1; FNIII: fibronectin Type III domains; N: short amino-terminal cytoplasmic tail; NCAM: neural cell adhesion molecule 1; NELL-1: neural epidermal growth factor 1; PLA2R: phospholipase A2 type M receptor; PSI: plexin-semaphorin-integrin; SEMA: semaphorin; SEMA 3B: semaphorin 3B; THSD7A: thrombospondin-type 1 domain containing 7A; TSD: ; TM: transmembrane domain; TSPN: thrombospondin-1-like molecule; VWC: von Willebrand factor Type C domains.
EXOSTOSIN 1/EXOSTOSIN 2-ASSOCIATED MEMBRANOUS NEPHROPATHY
Examining with the aforementioned techniques both serum samples and glomerular eluates in patients with MN negative for PLA2R, the first antigens found were EXT1/EXT2. The GBM is principally composed of Type IV collagen, laminin, nidogen, and heparin sulfate proteoglycans. Agrin and perlecan are the main heparin sulfates in GBM. Heparin sulfates are present in the GBM, matrix, and cell surfaces.
EXTs are glycosyltransferases that provide the synthesis of the heparin sulfate, adding glycosaminoglycan residues to the core protein with the generation of complex polysaccharides.18,19 EXT1/EXT2 are a heterodimeric enzyme called glycosyltransferase, which adds glycosylic residues to the protein backbone of proteoglycans.20 They are transmembrane proteins situated in the endoplasmic reticulum where biosynthesis of the heparin sulphates occurs. EXT proteins have a short amino terminal cytoplasmic tail; a single transmembrane domain; a stem region; and a long, globular catalytic carboxyl-terminal domain within the Golgi lumen.21,22 EXTs are secreted into the extracellular medium in a truncated form.23 The heterodimer of EXT1/EXT2 shows structural similarities and can exist as heterodimers that have increased stability and activity.24 Mutations in EXT1/EXT2 cause the autosomal dominant disorder hereditary multiple exostoses.25,26
Frequently, examining serum samples by Western blot does not allow the detection of circulating anti-EXT1/EXT2 antibodies. There are two possibilities for this: the antibodies could not be detected because they are directed to specific epitopes of the truncated protein that are absent in the recombinant EXT1/EXT2 protein; or it is possible that serum antibodies could not be detected because their titre is very low.
EXT1/EXT2-MNs are more present in females, and more than 70% of patients have abnormal laboratory values for antinuclear antibodies, anti-double stranded DNA, anti-Smith antibodies, or anti-Sjögren’s syndrome-related antigen A or B.27 34% of patients have a clinical diagnosis of systemic lupus erythematosus. The antibodies present in the GBM are predominantly IgG1, and the classical pathway activates the complement.
NEURAL CELL ADHESION MOLECULE 1
Neural cell adhesion molecule (NCAM-1) is frequently found in membranous lupus nephropathy, but may also be observed in primary MN as well. NCAM-1 is a member of the Ig superfamily of proteins of 150 kDa molecular weight. NCAM-1 was identified by Caza et al.,16 adding immunoprecipitation on frozen biopsies to the techniques of Sethi et al.13 NCAM-1 contains an amino-terminal tail, five immunoglobulin domains (IgG), two fibronectin Type III domains, a transmembrane domain, and an intracellular region at the C-terminal end. In adults, NCAM-1 may be found at high levels in different organs and tissue as the central nervous system, and cells within the immune system. Neuropsychiatric disorders occurred in 40% of patients who are NCAM-1 positive, probably due to NCAM-1 high expression in the central nervous system.28 NCAM-1 could not be found in normal kidneys, while it was found in urinary exosomes.29 In patients with lupus nephritis-related MN, NCAM-1 levels in the urine correlated with disease activity.30 It is possible to find antibodies directed against NCAM-1 in the patient’s sera. NCAM-1 is responsible for MN in many patients with lupus nephropathy, where it activates the complement by the classical pathway. In conclusion, NCAM-1 ranks second after EXT1/EXT2 in the list of antigens in membranous lupus nephropathy.
NEURAL EPIDERMAL GROWTH FACTOR-LIKE 1 PROTEIN
Neural epidermal growth factor 1 (NELL-1) is a secreted, 90 kDa protein that promotes bone regeneration.31 Patients with craniosynostosis have high levels of NELL-1. In pathological conditions. NELL-1 expression is higher in tubules, and from 5–25% of glomerular cells express NELL-1 at the mRNA level.32 NELL-1 is a cytoplasmic protein kinase C-binding protein. Its structure contains an amino-terminal thrombospondin-1-like molecule, coiled-coil domain, four von Willebrand-type domains, and six epidermal growth factor-like repeats.33
Sethi et al.15 selected patients with PLA2R negative MN, and by laser microdissection and MS identified NELL-1. By immunohistochemistry a granular anti-NELL-1 GBM staining was documented, and by confocal microscopy there was a colocalisation of NELL-1 and IgG. By Western blot, serum antibodies to NELL-1 were detected. NELL-1 is probably localised in the podocytes, and does not derive from circulating antigens or immune complexes.34
Kudose et al.35 examined more than 2,000 non-lupus MNs. 50% of them showed segmental membranous glomerulonephritis defined by subepithelial deposits involving 50% of the GBM. Among these segmental membranous glomerulonephritis, NELL-1 staining was present in 25%. According to this study, NELL-1 appears to be the first antigen in MN.
NELL-1-associated MN appears as a primary MN. However, in some cases, malignancy may be associated. According to Caza et al.,36 the incidence of malignancy varies from 10% to 33%.
PROTOCADHERIN 7-ASSOCIATED MEMBRANOUS NEPHROPATHY
Protocadherin-7 (PCDH7) is another recently discovered antigen related to MN. Sethi et al.37 examined PCDH7 with the usual techniques, and after laser microdissection kidney biopsies, and sera of 135 patients with PLA2R negative MN. In addition, immunohistochemistry, immunofluorescence, and confocal microscopy were used to confirm the MS/MS findings. Two validation cohorts38 validated all the new findings. Immunohistochemistry showed a bright granular staining along the GBM. PCDH7 and IgG were colocalised, and Western blot analysis documented autoantibodies to PCDH7.
Cadherins are a large group of transmembrane proteins that mediate cell-cell recognition and adhesion.39 Their function is unknown, but is probably important in cell signalling.40
In conclusion, PCDH7 has been identified as a new antigen, and autoantibodies against PCDH7 were present in adult patients with PLA2R negative MN. PCDH7 is mostly present in older patients, complement activation is minimal, and spontaneous remission without immunosuppressive treatment is frequent. Complement activation in PCDH7-associated MN was lower when compared with other antigen associated MNs.41
After PCDH7 discovery, the new classification of MNs is reported in Figure 3.
The overall incidence of primary MN that the studies reported in the beginning ranged approximately 70%, are to date about 80%. This is due to the discovery of the new antigens described. In addition to the different percentage reported in Figure 3, it is interesting to examine several studies that have been made on the specificity and sensitivity of the new antigens and relative biomarkers.
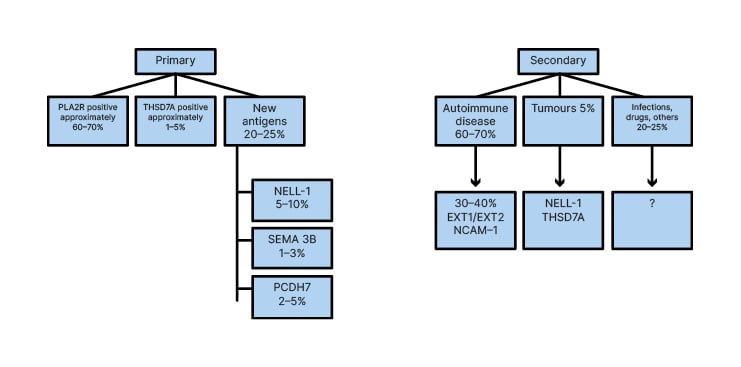
Figure 3: New and recent classification of membranous nephropathies.
EXT1: exostosin 1; EXT2: exostosin 2; NCAM-1: neural cell adhesion molecule 1; NELL-1: neural epidermal growth factor 1; PCDH7: protocadherin-7; PLA2R: phospholipase A2 type M receptor; SEMA 3B: semaphoring 3B; THSD7A: thrombospondin-type 1 domain containing 7A.
In an initial study based on PLA2R, IgG4 and THSD7A,42 the diagnostic performance of the three biomarkers reached 79% of sensitivity and 83% of specificity. A recent study43 carefully examines all the MN-related podocyte target antigen and autoantibody biomarkers, but does not face the issue of their specificity and sensitivity. A different study44 explores the differences in molecular mechanisms and key biomarkers between membranous nephropathy and lupus nephritis. According to this study, the best diagnostic value in differentiating patients with MN from patients with lupus nephritis is of the genes IFI44 and MX1, with an area under the curve of 0.980 and 0.967, respectively, while NELL-1 has no diagnostic value.
In a very recent study by Sealfon et al.,45 the authors examined in the area under the curve the accuracy in distinguishing genes of MN from those of other nephropathies. The authors found that MN classifier has the highest accuracy across different diseases in two independent cohorts.
MORE ANTIGENS ON THE HORIZON
Other antigens will be discovered soon or in the future, as no antigen has been identified in 10–20% of primary MNs. Just recently, at last three new antigens have been discovered, as well as their mechanism of action.
Xu et al.46 found in a male patient with MN-associated neurological disorders the presence of autoantibodies against paranodal proteins such as neurofascin 155 (NF155) and contactin-1 (CNTN1). Reviewing the literature, the authors found 22 cases of chronic inflammatory demyelinating polyneuropathy,47 five of which were anti-CNTN1 antibody-associated.48-51 Debiec et al.52 clarified the pathophysiology of this neuro-renal syndrome.
CNTN1 and NF155 represent a structure in the paranode that improves cell adhesion and receptor stabilisation. IgG4 autoantibodies block protein-protein interactions between NF155 and CNTN1, and this facilitates the paranodal terminal loop detachment from the axon. This determines a severe nerve conduction failure that represents a severe disease, also without inflammation and complement cascade activation.
On the other hand, similar immune complexes are found formed at the basal side of the podocyte. As always, such glomerular subepithelial IgG4 deposits may be due either to an in situ formation of immune complexes, or to the deposition of preformed circulating immune complexes, both including the complement activation.
Caza et al.53 recently found a Type III TGF-β receptor (TGFBR3) as a new biomarker that is present in some patients with membranous lupus nephritis. After mass spectrometry and laser microdissection, confocal microscopy was used to find colocalisation of immune complexes and IgG. TGFBR3 was found principally in glomeruli and coprecipitated with IgG. TGFBR3 colocalised with IgG along the GBM in TGFBR3-associated MN. In conclusion, a diagnosis of TGFBR3-associated MN should raise the suspicion for a concomitant autoimmune disease.
The high temperature recombinant protein A1 (HTRA1) is a new target antigen recently found in primary MN by Al-Rabadi et al.54 HTRA1 is a serine protease identified as a new podocyte antigen in some patients with primary MN. At immunoblotting, sera from two patients reacted either with a 51 kDa protein within glomerular extract or with recombinant human HTRA1. Anti-HTRA1 antibodies were predominantly IgG4. In the course of active disease, the patients’ sera revealed significantly higher titres of anti-HTRA1 antibody than the sera collected during the remission phase. HTRA1 was principally detected within immune deposits of HTRA1-associated MN in 14 patients identified among three different cohorts. The analysis of 118 patients who were MN-negative for any known antigen documented a prevalence of 4.2% for HTRA1.
CONCLUSION
Since the beginning of the studies, membranous nephropathy has been distinguished into two different categories: primary or idiopathic when it is not associated with other diseases; and secondary when it is associated with diseases such as tumours, infections, or autoimmune disorders. It should be highlighted that the pathological aspects do not allow a distinction between the two forms. Primary MN is to be ascribed to an autoimmune disorder where autoantibodies are directed against antigens represented by proteins of the GBM. The first antigen discovered was NEP. The disease was discovered in newborns born to females who were pregnant who develop antibodies because of a deficiency of NEP due to mutations in the NEP gene. The majority of primary MN have as target antigens either PLA2R or THSD7A.
These two antigens account for the majority of primary MN, but not for all. New techniques principally introduced by Sethi et al.13 allowed identifying other less frequently involved antigens. As has been described in this study these antigens are EXT1/EXT2, NELL-1, SEMA 3B, PCDH7, and NCAM-1. More recently, other antigens have been discovered as contactin associated with neurological disturbances, TGFBR3, and HTRA1. Almost certainly, other antigens responsible for primary MN will be discovered in the future. As already mentioned, the histological pattern is similar for all the primary MNs, even if each of them is characterised by different clinical and outcome findings. On this basis, in a very recent study, Sethi et al.55,56 propose that any of the protein/antigen-associated MN represents a specific disease, whose histological renal aspects are similar and characterised by subepithelial immune depositions on the GBM.