Abstract
Autosomal dominant polycystic kidney disease is the most common inherited kidney disease and results from mutations in the polycystin 1 gene (PKD1) or the polycystin 2 gene (PKD2). The disease is characterised by the progressive development of fluid-filled cysts derived from renal tubular epithelial cells that destroy the architecture of the renal parenchyma and lead to kidney failure. Until recently, the causes and the molecular pathways that lead to cystogenesis remained obscure. In the last decade, enormous progress has been made in understanding the pathogenesis of autosomal dominant polycystic kidney disease and developing new therapies. The purpose of this review is to provide an update on the promising therapies that are being developed and tested, based on knowledge of recent advances in molecular and cellular targets involved in cystogenesis.
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) affects 1:400–1:1,000 of live births and is the most common monogenic inherited form of kidney disease across all ethnic types. ADPKD is characterised by cyst formation and enlargement in the kidney and other organs. It represents the fourth leading global cause for kidney failure, and end-stage renal disease (ESRD) usually occurs by late middle age, requiring renal replacement therapy in ~50% of patients by 70 years of age.1,2
In 85% of cases, ADPKD occurs as a result of germ-line mutation in the polycystin 1 gene (PKD1), while in 15% of cases it is due to a germline mutation in the polycystin 2 gene (PKD2).3 Recently, a polycystic kidney and/or polycystic liver disease-3 (PKD3) caused by heterozygous mutation in the gene encoding for glucosidase II subunit-a (GANAB) has been described.4 Polycystin-1 (PC1) and polycystin-2 (PC2) interact with each other through their C-terminal cytoplasmic domains and are known to form a complex that functions as a transient receptor potential channel involved in the regulation of intracellular calcium homeostasis.5,6 Glucosidase II subunit-a is required for maturation and surface and ciliary localisation of PC1 and PC2.
Analysis of GANAB-null human renal cells resulted in absence of the mature N-terminal PC1 but full-length PC1 and PC2. Heterozygous-null CANAB renal cells had a 50% depletion of mature N-terminal PC1.4 On average, patients with mutations in PKD1 developed ESRD at younger ages.7
Cystogenesis follows a two-hit model. ADPKD is recessive at the cellular level and cysts develop clonally from a tubular cell only after the cell has acquired a second, somatic mutation to inactivate the remaining normal allele.8 Although the exact mechanisms of cystogenesis remain to be elucidated, the pathological processes that facilitate cyst enlargement are probably a result of two specific abnormalities: i) increased fluid secretion into the cyst lumen, and ii) inappropriately increased cell division by the epithelium lining the cyst.9 The major signalling pathways implicated in these phenotypic changes include the intracellular deregulation of calcium homeostasis, cyclic adenosine monophosphate (cAMP) accumulation and activation of protein kinase A (PKA), activation of mitogen activated protein and mammalian target of rapamycin (mTOR) kinases, and other intracellular signalling mechanisms.10-12
Until recently, the treatment of ADPKD was aimed at the management of secondary conditions, particularly hypertension, to limit morbidity and mortality after the disease becomes symptomatic. Recent developments arising from a better mechanistic understanding of the molecular pathways involved in cyst growth have allowed targeting the disease pathogenesis, rather than the disease complications. The current review focusses on these novel therapeutic approaches that interfere with the molecular pathways of cystogenesis (Figure 1).
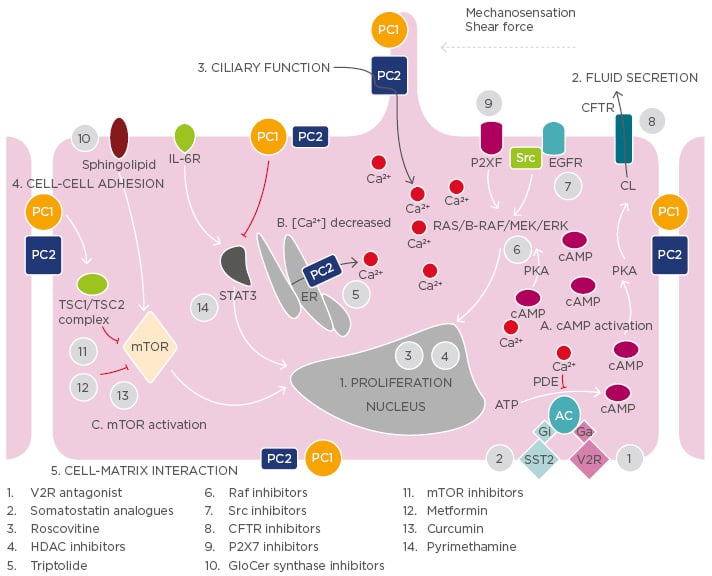
Figure 1: Illustration of the key mechanisms of adult autosomal polycystic kidney disease pathogenesis and targets of potential treatments.
Polycystin-1 and polycystin-2 expressed in different subcellular locations and regulate 1) proliferation, 2) fluid secretion, 3) ciliary function, 4) cell-cell adhesion, and 5) cell-matrix interaction of renal epithelial cells. Dysfunction of polycystin-1 or polycystin-2 results to aberrant signalling pathways including: A) activation of cAMP, B) decreased intracellular calcium concentrations, and C) activation of mTOR.
The targets of candidate drugs are depicted as grey circles.
CFTR: cystic fibrosis transmembrane regulator; ER: endoplasmic reticulum; ERK: extracellular-signal
regulated kinase; GlcCer: glucosylceramide; HDAC: histone deacetylase; IL-6R: interleukin-6 receptor; MEK: mitogen activated protein kinase; mTOR: mammalian target of rapamycin; PC: polycystin;
PDE: phosphodiesterase; PKA: protein kinase A; SR: somatostatin receptor; TSC: tuberous
sclerosis; V2R: vasopressin V2 receptor; EGFR: estimated glomerular filtration rate; cAMP: cyclic
adenosine monophosphate.
DRUGS TARGETING cAMP-DEPENDENT CYSTIC EXPANSION
Role of cAMP in Cystogenesis
In ADPKD, disruption of intracellular Ca2+ homeostasis due to mutations in the PKD gene leads to low intracellular calcium and consequently increased levels of intracellular cAMP. Normally, the levels of cAMP are controlled by a balanced activity of membrane-bound and soluble isoforms of adenylate cyclase (AC) (which catalyses the formation of cAMP from ATP) and phosphodiesterases (which degrades cAMP to AMP). Decreased intracellular calcium inhibits the activity of phosphodiesterases and activates ACs, thus producing a net increase in cAMP concentration.10 cAMP exerts its effects via PKA, which phosphorylates a number of metabolic enzymes and promotes transepithelial fluid secretion. Chloride secretion drives sodium into the cystic cavity through paracellular mechanisms; this causes movement of water through aquaporins and cyst expansion.13 In addition, in ADPKD, cAMP promotes cyst enlargement by stimulating epithelial cell proliferation, primarily through the activation of the B-Raf/MEK/ERK pathway.14,15
Vasopressin 2 Receptor Antagonists
Normally, vasopressin (AVP) is secreted into the circulation by the posterior pituitary gland, in response to an increase in serum osmolality or a decrease in effective circulating volume. In the kidney, AVP binds to the V2 AVP receptor in the basolateral membranes of collecting-duct cells. The V2 receptor is a typical member of the large superfamily of G protein–coupled receptors. Thus, occupancy of this receptor results in mediated activation of AC and the formation of cAMP with subsequent activation of PKA, which promotes the fusion of cytoplasmic vesicles containing aquaporin-2 water-channel proteins with the apical membrane. As a result, this normally water-tight membrane becomes water-permeable. Driven by the osmotic gradient of sodium, water is then transcellularly reabsorbed, entering the cells through aquaporin-2 in the apical membrane and leaving the cells for the interstitial space through aquaporin-3 and aquaporin-4, which reside in the basolateral membrane.16 In patients with ADPKD, there is a pathologically hyperactive AVP/V2 receptor system. Serum concentrations of AVP correlate positively with both serum osmolality, as well as with total kidney size and negatively with glomerular filtration rate (GFR).17-20 The central role of cAMP in cystogenesis and the pathologically hyperactive AVP/V2 receptor system have made the blocking of V2R particularly appealing in the treatment of ADPKD.
In preclinical trials, a non-peptide AVP antagonist mozavaptan (OPC-31260), administered in murine cystic models orthologous to human disease, including the Pkd2WS25/− mouse (ADPKD), PCK rat (ARPKD), and pcy mouse (nephronophthisis Type 3), reduced renal cAMP and inhibited disease progression, as measured by the reduction in kidney volume, the cystic area, the number of mitotic and apoptotic cells, and the blood urea nitrogen.21-23 Additional studies were conducted to examine the effects of tolvaptan (OPC-4106), a more potent and highly selective human V2R antagonist, in comparison with mozavaptan.24 Tolvaptan showed similar results on renal cAMP and PKD progression in the PCK rat model using the lowest dose. Reif et al.25 in an in vitro study examined the effect of tolvaptan on intracellular cAMP, ERK activity, cell proliferation, and transcellular chloride anion secretion using human ADPKD cyst epithelial cells. Tolvaptan caused inhibition of cAMP AVP-induced production, ERK signalling AVP-induced, cell proliferation, and chloride anion secretion. These effects significantly contributed to decreased in vitro cyst growth.
The large randomised, double-blind, placebo-controlled, multinational, Phase III TEMPO 3:4 trial26 confirmed the aforementioned experimental studies. This trial enrolled 1,445 patients aged 18–50 years with ADPKD, rapidly progressive kidney growth (total kidney volume [TKV] ≥750 mL) as measured by magnetic resonance imaging (MRI) and chronic kidney disease (CKD) Stages 1–3. Tolvaptan reduced the rate of TKV growth (primary endpoint) by 49% and the rate of estimated GFR (eGFR) loss on treatment (secondary endpoint) by 26% per year during the median observation period of 3 years. The effect on TKV appeared greater during the first year of treatment than during the second or third years. Beneficial effects on renal function have been observed in all patient subgroups, especially in patients aged ≥35 years and in patients with hypertension or a TKV of ≥1,500 mL. Another important secondary endpoint was the reduction in kidney pain occurring early and throughout treatment. The results of the TEMPO 3:4 trial suggested that tolvaptan had no effect compared with placebo on albuminuria. Conversely, in a post hoc exploratory analysis, tolvaptan decreased albuminuria compared with placebo independently of blood pressure. In addition, the treatment efficacy of tolvaptan on changes in TKV and eGFR was more readily detected in patients with higher albuminuria.27
Based on the results of the TEMPO 3:4 trial, tolvaptan has been approved to delay the progression of ADPKD in patients with a rapid increase of TKV in Japan, Canada, the European Union (EU), the UK, and South Korea. The European Renal Association-European Dialysis and Transplant Association (ERA-EDTA)28 has issued detailed guidance on this topic. ERA-EDTA suggests that tolvaptan can be prescribed to adult ADPKD patients aged <50 years with CKD Stages 1–3a (eGFR >45 mL/min/1.73 m2) who have demonstrated, or who are likely to have, rapidly progressing disease. ERA-EDTA recommends not starting tolvaptan in patients aged 30–40 years with CKD Stage 1 (eGFR >90 mL/min/1.73 m2) or patients aged 40–50 years with CKD Stages 1 or 2 (eGFR >60 mL/min/1.73 m2). The organisation recommends that rapid disease progression be defined as a confirmed annual eGFR decline of ≥5 mL/min/1.73 m2 in 1 year, and/or ≥2.5 mL/min/ 1.73 m2 per year over a period of 5 years. It can also be defined as a >5% increase in TKV per year by repeated measurements (preferably ≥3, each at least 6 months apart and by MRI). With regard to dosing, ERA-EDTA suggests that tolvaptan be started with a dose of 45 mg in the morning and 15 mg in the evening, uptitrating the dose to 50/30 and 90/30 when tolerated, and discontinuing tolvaptan when patients approach ESRD.
Tolvaptan has significant adverse effects including aquaretic effects (polyuria, nocturia, and polydipsia) and elevation of aminotransferase enzyme concentrations with the potential for acute liver failure.26,29,30 Although the incidence of hepatic enzyme elevation was low, three patients treated with tolvaptan in the TEMPO 3:4 trial met criteria for Hy’s law (elevation of aminotransferase enzymes >3-times the upper limit of normal, plus elevation of total bilirubin >2-times the upper limit of normal, without other explanatory mechanisms), which have a fatality rate of ˜10% from liver injury. Therefore, the Cardiovascular and Renal Drugs Advisory Committee of the US Food and Drug Administration (FDA) declined to approve tolvaptan for ADPKD, as they were worried that liver damage might progress with long use treatment.31 Appropriate patient selection is critical to optimise long-term benefits and minimise adverse effects and hepatotoxic risk factors.
Studies to further assess the efficacy and tolerability of tolvaptan in patients with ADPKD are ongoing or just completed. TEMPO 4:4 is a 2-year, open-label extension of TEMPO 3:4 and was completed in March 2016. This study aimed to evaluate the long-term efficacy and safety of tolvaptan in patients with ADPKD; the findings are soon to be published. In addition, the long-term safety of titrated tolvaptan in patients with ADPKD is currently being assessed in a Phase III open label trial,32 while the ongoing Phase IIIb REPRISE trial aims to extend the understanding of the efficacy and safety of tolvaptan in patients with late Stage 2–early Stage 4 CKD.33
Somatostatin Analogues
Somatostatin (SST) is an endogenous hormone primarily secreted by the pancreatic islet δ-cells. SST has anti-secretory and anti-proliferative effects mediated by the interaction with five subtypes of G protein–coupled receptors (SSTR1-5).34 SST receptors are expressed by renal tubular epithelial cells and by cholangiocytes. SST selectively inhibits cAMP synthesis in the epithelial cells of the distal tubules and collecting ducts both in vitro and in vivo35,36 and exerts similar effects to cholangiocytes.37 As plasma half-life of the native SST is very short (1–3 minutes), the synthetic analogues octreotide, lanreotide, and pasireotide were developed for use in clinical practice.
In particular, octreotide and lanreotide have a half-life of 2 hours and present a high affinity for SSTR2 and SSTR3 and moderate affinity for SSTR5. By comparison, pasireotide has high affinity for all the receptors of SST, except SSTR4, and its plasma half-life is about 12 hours.38 Currently, formulations of octreotide and lanreotide with long-acting release (LAR), which allows the administration every 28 days intramuscularly or intradermally, have been introduced into clinical practice. Ruggenenti et al.39 have evaluated for the first time the effectiveness of octreotide-LAR by performing a randomised, crossover, placebo-controlled trial in 14 ADPKD patients, which demonstrated the potential efficacy in slowing the growth of TKV and the relative safety of the treatment. Van Keimpema et al.40 compared the effects of 6 months of treatment with lanreotide or placebo in 54 patients with polycystic liver disease (PLD), including 32 with ADPKD and the remaining with isolated PLD. The average volume of the liver decreased in patients treated with lanreotide while it increased in the placebo group. Moreover, in patients with ADPKD, TKV was reduced after treatment with lanreotide, while it was increased in the placebo group. In a subsequent open label extension study41 patients who participated in the initial trial were re-enrolled to complete a treatment period of 12 months with lanreotide. Liver volume decreased after 12 months of treatment with lanreotide, with the greatest effect seen during the first 6 months. In the 25 patients with ADPKD, TKV remained stable at the end of 12 months. In another 12-month study, 42 patients with PLD, including 34 with ADPKD, were randomised to receive treatment with octreotide-LAR or placebo.42 The total volume of the liver was reduced in the treatment arm with octreotide-LAR but increased in the placebo group. In patients with ADPKD, the TKV remained unchanged in the octreotide-LAR group but increased in the placebo group. In addition, renal function had a slower reduction in patients treated with octreotide-LAR, although the difference did not reach statistical significance. More recently in the ALADIN multicentre study conducted in Italy, 79 patients with ADPKD and eGFR >40 mL/min/1.73 m2 were randomised to a 3-year treatment with octreotide-LAR or placebo.43 After the first year, the average increase in TKV was significantly lower in patients treated with octreotide-LAR compared to those receiving placebo. In the third year, the average increase in TKV in the treatment arm was lower than the placebo group without reaching statistical significance. During the entire study period, the annual reduction in GFR was lower in the octreotide-LAR group than in the placebo group, although the difference did not reach statistical significance. A more recent open label clinical study evaluated the efficacy of 6 months of treatment with lanreotide in 43 patients with symptomatic PLD and ADPKD (eGFR >30 mL/min/1.73 m2).44 Compared to baseline, the median liver volume decreased significantly, as well as that of the kidney. In addition, renal function remained stable until the end of the study. A recent meta-analysis confirmed the efficacy of SST analogues in reducing the progressive increase of TKV on average, with a reduction of 9% compared to the growth observed in patients treated with placebo or conventional therapies. However, treatment with SST analogues did not demonstrate significant effects on the eGFR.45
Based on these studies, in August 2015 the European Medicines Agency (EMA) has attributed to lanreotide the ‘orphan drug’ designation for the treatment of ADPKD. Designated orphan medicinal products are products that are still under investigation and are considered for orphan designation on the basis of potential activity. Opinions on orphan medicinal product designations are based on the following three criteria: i) the seriousness of the condition; ii) the existence of alternative methods of diagnosis, prevention, or treatment; and iii) either the rarity of the condition (affecting no more than 5 in 10,000 people in the EU) or insufficient returns on investment.
In the studies mentioned above, treatment with SST analogues was generally well tolerated with no particular problems, diarrhoea being the most common adverse event. However, recently, the authors of a randomised, controlled trail documented an increased risk for hepatic cyst infection during lanreotide treatment. A literature review also suggested an increased risk for hepatic cyst infection during the use of SST analogues.46
Additional clinical trials of SST analogues for ADPKD and/or PLD are currently ongoing.47-50
DRUGS TARGETING THE mTOR SIGNALLING PATHWAY
Role of the mTOR Signalling Pathway in Cystogenesis
Serine/threonine-protein kinase mTOR is an enzyme that plays a critical role in proliferation and cell growth.51 The first suggestion of a prominent role of the mTOR pathway in the pathogenesis of ADPKD comes from studies in patients with severe infantile-onset of ADPKD due to a large deletion of chromosome 16 involving the PKD1 gene [16p13.3] and the adjacent tuberous sclerosis 2 (TSC2) genes [16p13.3].52 TSC1 and TSC2 encode for hamartin and tuberin, respectively. These two proteins together with TBC1 domain family member 7 (TBC1D7) form the TSC protein complex that acts as a critical negative regulator of mTOR complex 1 (mTORC1).53 PC1 has also an important function in the regulation of the mTOR pathway, as the C-terminal cytoplasmic tail of PC1 interacts with tuberin. In ADPKD this interaction is impaired, and the mTOR pathway is inappropriately activated in cyst-lining epithelial cells of human ADPKD patients and mouse models.54 Based on these data, a possible therapeutic role for mTOR inhibitors in ADPKD has been suggested.
mTOR Inhibitors
Sirolimus and its derivative everolimus, used in maintenance immunosuppression in patients undergoing kidney transplantation, have been proposed as potential new drugs to slow the growth of cysts and the progression of ADPKD in ESRD. The effects of treatment with mTOR inhibitors have been assessed in different experimental models of ADPKD.55-58 The first published randomised double-blind study compared the effects of 2 years of treatment with everolimus (5 mg/day) or placebo in 433 patients with ADPKD and GFR >30 mL/min/1.73 m2.59 During the first year of study, the increase of TKV was significantly lower in the treatment arm with everolimus compared to placebo. This effect was not confirmed at the end of the second year. In addition, the initial effectiveness of everolimus in slowing TKV did not translate into improvement of renal function.
The SUISSE study compared the effects of treatment for 18 months with sirolimus (2 mg/day) or conventional therapy in 100 patients with ADPKD and GFR ≥70 mL/min/1.73 m2.60 The median increase in TKV was comparable between the two groups as well as the eGFR throughout the entire study period. The randomised trial SIRENA compared the effects of treatment with sirolimus or with conventional therapy alone for 6 months in 21 patients with ADPKD and GFR ≥40 mL/min/1.73 m2.61 The treatment with sirolimus was associated with a minor increase of the TKV compared to conventional therapy. In a subsequent open label study (RAPYD), 55 patients with ADPKD and mild-to-moderate renal impairment were randomised to 24 months of treatment with ramipril (control group), ramipril in combination with high doses of sirolimus (target blood levels: 6–8 ng/mL) or ramipril in combination with low-dose sirolimus (target blood levels: 2–4 ng/mL).62 Compared to baseline, total cyst volume decreased significantly in both treatment arms with sirolimus, while increasing in the control group. In a more recent study, 30 patients with ADPKD and measured GFR ≥25 mL/min/1.73 m2 were randomised to receive low-dose sirolimus (target blood levels 2–5 ng/mL), standard doses of sirolimus (target blood levels >5–8 ng/mL) or conventional therapy for 12 months.63 TKV did not change significantly in the two treatment groups with sirolimus as in the group assigned to conventional therapy. In addition, the renal function improved (GFR measured by plasma clearance of iothalamate) with low-dose sirolimus but not with the standard dose of the drug. Currently, there are two ongoing trials that are testing mTOR inhibitors in ADPKD.64,65
OTHER THERAPEUTIC TARGETS IN PRECLINICAL STUDIES AND IN EARLY CLINICAL TRIALS
In addition, other agents targeting different molecules or pathways involved in cystogenesis have been used in preclinical studies and some of them are ongoing in early clinical trials in humans.66 All these agents are summarised in Table 1.
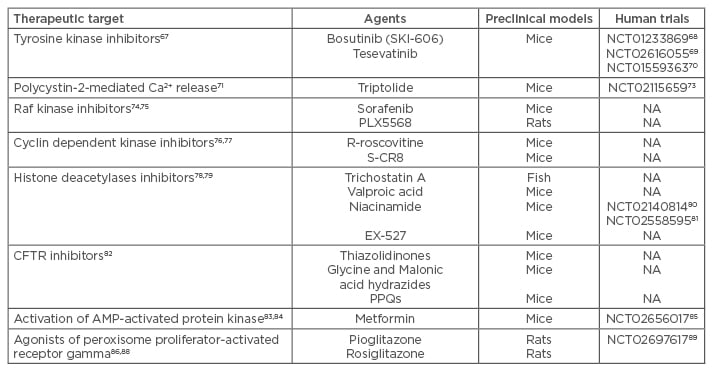
Table 1: New agents in preclinical models or in early clinical trials in humans.
CFTR: cystic fibrosis transmembrane conductance regulator; PPQs: pyrimido-pyrrolo-quinoxalinediones; AMP: adenosine monophosphate.
Bosutinib (SKI-606) is a Src/Abl tyrosine kinase inhibitor effective in inhibiting epithelial cell proliferation and reducing extracellular matrix adhesion. In the BPK and PCK rodent models of ADPKD, bosutinib was found to suppress kidney cyst formation by inhibiting epidermal growth factor receptor activation and downregulating B-RAF/ERK signalling.67 Based on this evidence, a Phase II, multicentre, randomised, double-blind, placebo-controlled clinical trial with bosutinib68 has been conducted and completed, and we currently are expecting publication of the study results. Tesevatinib, a new tyrosine kinase inhibitor, is being currently evaluated in two ongoing trials.69,70
Triptolide, by acting as a PC2 agonist to restore cytosolic Ca2+ release, has been effective in arresting cellular proliferation and attenuating overall cyst formation in Pkd1−/− murine kidney epithelial cells.71 A clinical trial conducted in China has been terminated due to a high rate of drop-outs72 and another trial is underway.73
In an experimental model, sorafenib, a non-selective RAF inhibitor, reduced the basal activity of ERK, inhibited cAMP-dependent activation of B-RAF and MEK/ERK signalling, and caused a concentration dependent inhibition of cell proliferation induced by cAMP and EGF. In addition, it completely blocked in vitro cyst growth of human ADPKD cystic cells.74 A different Raf inhibitor (PLX5568) has been evaluated in the Han:SPRD rat model.75 In this study, cyst enlargement attenuated without an improvement in kidney function. Furthermore, the authors reported increased renal and liver fibrosis.
A preclinical study with the CDK inhibitor (R)-Roscovitine in juvenile cystic kidney and congenital polycystic kidney mouse models of PKD effectively attenuated cystogenesis by inhibiting cell cycle progression, proliferation, and apoptosis.76 In addition, a more potent second-generation analogue of roscovitine (S-CR8) showed effective inhibition of both renal and hepatic cystogenesis in an orthologous mouse model of ADPKD with inactivated PKD1 gene.77
Altered expression of histone deacetylases (HDCAs) causes abnormal transcription of key genes controlling principal cellular functions such as cell proliferation, cell-cycle regulation, and apoptosis.78 A pan-HDAC inhibitor called trichostatin A (TSA) has been evaluated in a PKD2 zebrafish model showing the ability to suppress pronephric cyst formation.79 The same results have been obtained after administration of valproic acid (VPA), a Class I HDAC inhibitor.79 The NIAC-PKD1 trial80 has just been completed, and we are currently awaiting publication of the results. In addition, another trial, NIAC-PKD2,81 is currently recruiting participants.
Each of the three chemical classes of cystic fibrosis transmembrane conductance regulator (CFTR) inhibitors has been tested in PKD models: i) thiazolidinones, ii) glycine and malonic acid hydrazides, and iii) pyrimido-pyrrolo-quinoxalinediones. The best thiazolidinone, tetrazolo-CFTRinh-172, and the best glycine hydrazide, Ph-GlyH-101, were found to inhibit cyst formation and enlargement in Madin–Darby canine kidney cyst models and in PKD1 mice.82
AMP-activated protein kinase regulates cell growth via suppression of the mTORC1 pathway, by direct phosphorylation of the tumour suppressor TSC2 and Raptor (regulatory associated protein of mTOR).83 Recently, Takiar et al.84 showed in a PKD1 mice model, that metformin inhibited renal cystogenesis and caused a significant decrease in the cystic index by activating AMP-activated protein kinase and suppressing mTOR and CFTR. Currently, the TAME trial85 is recruiting ADPKD patients to see if metformin is safe and well tolerated.
Agonists of peroxisome proliferator-activated receptor gamma (PPAR-γ) have been shown to have anti-cystogenic properties in PKD animal models. Pioglitazone has been shown to inhibit the growth of renal and hepatic cysts in PCK rats by inhibiting the CFTR-mediated ionic current and the secretion of fluid.86 In another study, pioglitazone also reduced cellular proliferation, highlighted by a reduction in the number of cells positive for Ki67 (a proliferation marker) in the dilated tubules and in cysts from treated rats.87 Another powerful agonist of PPAR-γ, rosiglitazone, has been used to treat Han:SPRD rats. Rosiglitazone delayed the onset of renal failure but was associated with cardiac enlargement due to excessive renal sodium reabsorption.88 Based on these preclinical data, the PIOPKD trial89 is currently recruiting participants.
CONCLUSION AND FUTURE DIRECTIONS
Multiple signalling pathways are involved in cyst formation and progression, and studies of these signalling pathways have led to potential treatments for ADPKD. In this review, we have covered the successes obtained in recent years in understanding the pathogenesis of ADPKD and presented novel therapeutic strategies targeting molecular pathways of cystogenesis. V2R antagonists and SST analogues have been shown to safely slow kidney growth and protect renal function in patients with ADPKD, and represent the most well-characterised and promising candidate therapies to date. According to the results of the TEMPO3/4 study and registration by the EMA, tolvaptan seems to be the first choice drug. Some medical interventions successful in experimental models failed in clinical practice and others still need to be evaluated in clinical trials. It is possible that monotherapy may not be sufficient and that targeting multiple molecular pathways will be required to retard cyst growth and disease progression in the feature. Combination therapy is then the right direction of further clinical trials in order to find effective treatment. To date no association of drugs inhibiting cystogenesis has proven to be effective: further research is needed. The association of tolvaptan and angiotensin-converting-enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), and statins are reported as effective associations in a recent study.90