Abstract
Immune complex-mediated membranoproliferative glomerulonephritis (iMPGN) can develop in association with autoimmune diseases such as primary biliary cholangitis (PBC), a chronic cholestatic liver disease characterised by destruction of the small and medium-sized bile ducts. Although the pathogenesis cannot be clearly defined, iMPGN and PBC overlap through the activation of innate and adaptive immune cells and the production of proinflammatory mediators. In this report, the authors present a case in which iMPGN and PBC were diagnosed simultaneously.
Key Points
1. The aim of the treatment of membranoproliferative glomerulonephritis due to secondary causes is to treat the underlying primary cause; for which renal support treatment is also essential. Immune complex-mediated membranoproliferative glomerulonephritis (iMPGN) secondary to primary biliary cholangitis (PBC), an autoimmune disease, is a very rare condition and its treatment is that of the secondary cause, PBC.
2. This is a very rare case report describing the association between iMPGN and PBC responding to ursodeoxycholic acid and non-immunosuppressive supportive therapy.
3. Every patient with iMPGN should be investigated for secondary aetiologies, and it should be kept in mind that, with appropriate treatment, iMPGN can be treated without the need for immunosuppressive therapy.
INTRODUCTION
Membranoproliferative glomerulonephritis (MPGN) is a glomerular disease characterised by mesangial expansion, endocapillary proliferation, and double contours of the glomerular basement membrane on light microscopy; by complement deposition with or without immune complex deposits on immunofluorescence; and by electron-dense deposits in the mesangium, subendothelium, or subepithelium on electron microscopy. Depending on the pathogenesis, MPGN is divided into complement-mediated MPGN and immune complex-mediated MPGN (iMPGN). iMPGN may develop secondary to chronic infections such as hepatitis B virus, hepatitis C virus, and monoclonal gammopathies, or in association with autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, or primary biliary cholangitis (PBC). Immune complexes, formed by the binding of antigens to antibodies in the bloodstream or in situ, can activate complement pathways, induce inflammatory cytokines, recruit leukocytes, and cause tissue damage.1
PBC is a chronic liver disease that mainly affects middle-aged females and is prevalent worldwide. The unclear aetiopathogenesis is thought to be due to a complex interaction between genetic factors such as HLA-DR8, and environmental factors such as microbial infections. The main histopathologic feature of PBC is chronic, non-suppurative destructive cholangitis leading to progressive loss of intrahepatic bile ducts and cholestasis. The most common symptoms are fatigue and pruritus. Other symptoms include jaundice, xanthomas, osteoporosis, portal hypertension, and hepatic encephalopathy.2 The association between MPGN and PBC is rare and has only been described in a few case reports.3,4 Here the authors present a case diagnosed with iMPGN and PBC, with parallel clinical courses of liver and kidney disease.
CASE PRESENTATION
A 43-year-old female patient with no medical history presented with complaints of fatigue, itching, jaundice, and oedema. She was not taking any medication. There was no family history of autoimmune diseases. The patient was referred to the nephrology department because of a serum creatinine level of 3.13 mg/dL (reference value: 0.5–0.9 mg/dL) and macrohematuria. The baseline creatinine value was not known. The daily urinary protein excretion rate was 10,400 mg/day (reference value: 80–150 mg/day). Viral (hepatitis B, hepatitis C, Epstein–Barr virus, HIV, rubella virus, rubeola virus, varicella, cytomegalovirus, and herpesvirus) and autoimmune markers (including rheumatoid factor, antiphospholipid IgM and IgG, the extractable nuclear antigen panel, anti-liver kidney microsomal, anti-double stranded DNA, antineutrophil cytoplasmic antibodies, antiglomerular basement membrane, and anti-smooth muscle) were all negative. Protein electrophoresis for monoclonal gammopathy of renal significance (MGRS) did not reveal any evidence of monoclonal gammopathy or light chain disease. Serum C3 level was 0.16 g/L (normal range: 0.79–1.52 g/L), and C4 level was 0.27 g/L (normal range: 0.16–0.38 g/L). An infectious process could not be detected by acute phase reactants, culture examinations, and imaging studies. Ultrasound-guided percutaneous renal biopsy revealed iMPGN with diffuse endocapillary and focal extracapillary proliferation. Fibrinoid necrosis was present, and neutrophils were noted to be a significant component of the endocapillary hypercellularity, resulting in an exudative picture. Double contours were identified along the glomerular basement membrane. Immunofluorescence staining showed diffuse global distribution of 3+ fine-coarse granular IgG and IgA deposits in the mesangium and along the peripheral capillary loops (Figure 1). Κ and λ were both positive; C3, IgM, and C1q were negative. Parallel studies revealed elevated levels of transaminases, alkaline phosphatase, IgM, and IgG. Itching was of great concern.
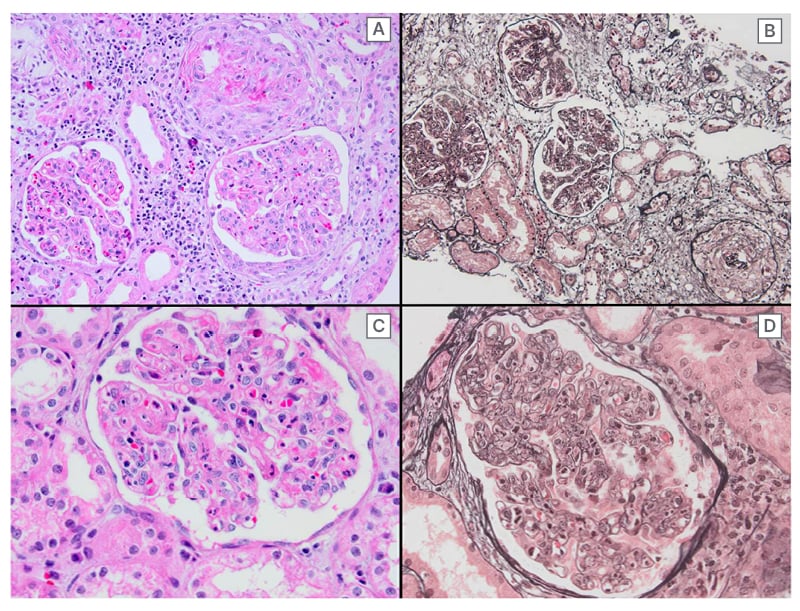
Figure 1: Immunofluorescence staining showing diffuse global distribution of 3+ fine-coarse granular IgG and IgA deposits in the mesangium and along the peripheral capillary loops.
Diffuse endocapillary and focal extracapillary proliferation can be discerned at medium power magnification (A: hematoxylin-eosin, B: Jones methamine silver stain). Note the presence of fibrinoid necrosis (A) and an abundance of neutrophils (C). Wide-spread double contours of the glomerular basement membrane, a so-called tram-tracking appearance, can be discerned at high power magnification (D: jones methamine silver stain).
The anti-mitochondrial antibody (AMA) and anti-nuclear antibody test were positive, >1/320 and +4 positive, respectively, and the patient was diagnosed with PBC (Table 1). The patient was thought to have developed iMPGN secondary to PBC.
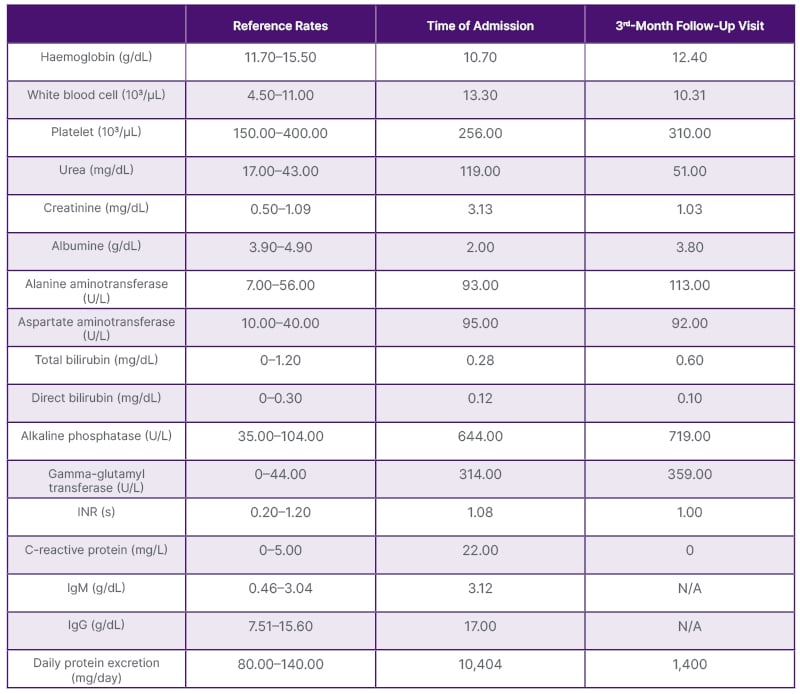
Table 1: Laboratory results at the time of admission and at the 3rd-month follow-up visit.
INR: international normalised ratio; N/A: not applicable.
Supportive treatments for nephrotic syndrome parameters, low-salt diet, ramipril 10 mg/day, and ursodeoxycholic acid (UDCA) 13 mg/kg/day were started. Outpatient follow-up was initiated. Gradually, liver enzymes, serum creatinine, and proteinuria decreased (Table 1).
DISCUSSION
In MPGN associated with autoimmune diseases, immune complexes can accumulate in the subendothelial or mesangial spaces of the glomerulus and cause glomerular damage and inflammation.4 Several hypotheses can be proposed for the development of MPGN during the course of PBC. Immune complexes containing AMAs or other autoantibodies may accumulate in the glomeruli and cause inflammation and damage.5 In addition, complement activation triggered by antigen-antibody complexes may lead to the formation of membrane attack complexes that damage the endothelium.6 Bile acids or other components of bile may facilitate the deposition of immune complexes by altering the permeability and charge of the glomerular basement membrane.7 Finally, bile epithelial cells, which are the target of autoimmune attacks in PBC, may damage the kidney with proinflammatory cytokines and chemokines, which may have endocrine as well as paracrine effects.8
In the management of MPGN due to secondary causes, treatment of the underlying primary cause and renal supportive therapy are essential. In the case presented here, the iMPGN has been thought to have developed secondary to PBC. Until the test results were available, attempts were made to control the complications of nephrotic syndrome and acute renal failure with supportive measures. When the diagnosis of PBC was confirmed, treatment with UDCA was started to improve bile flow and reduce oxidative stress.9 In the following period, renal function tests improved significantly under antiproteinuric therapy with ramipril 10 mg/day (maximum dose) and a low-salt diet. Immunosuppressive therapy was not considered necessary due to the improvement in renal function following the renoprotective therapy. UDCA can reduce the formation of immune complexes by inhibiting the production of proinflammatory cytokines and chemokines, reducing the activation and proliferation of T cells, inducing regulatory T cells, and modulating B cell function. In addition, UDCA can reduce the damage caused by immune complexes by inhibiting complement activation and leukocyte uptake.10
In the literature, the authors found only two cases in which the two diseases were associated with each other. In a case published by Koike et al.,3 a 49-year-old male patient with PBC and Sjoegren’s syndrome was diagnosed with a Type 3 MPGN-like lesion secondary to proteinuria, which was treated with prednisolone and supportive measures.3 In another study by Zand et al.4 on patients with MPGN having autoimmune diseases, a 57-year-old female patient with undifferentiated connective tissue disease and PBC was treated with azathioprine/prednisone in combination with supportive therapy.4 During follow-up, both patients were not on dialysis, as was the case with the authors’ patient. The patient’s first hospitalisation was when the authors took up the case.
For this reason, the baseline creatinine level could not be determined. The patient’s most serious complaint was itching but no infection was found in her medical history. No infectious processes were detected by acute phase reactants, culture examinations, and imaging studies. There was no C3, IgM, or C1q deposition on immunofluorescent microscopy. Lupus or IgA nephropathy were ruled out due to the presence of double contours. The authors suspected it to be glomerulonephritis with a membranoproliferative pattern of injury and IgG and IgA deposition, not only in the mesangium but also along the glomerular capillary walls. Morphologically, both post-infectious and infection-related glomerulonephritis would enter the differential diagnosis. When the results of histopathologic analysis were available, the evaluations were revisited as post-infectious glomerulonephritis was included in the differential diagnosis. However, no evidence of infection was found. When a detailed analysis revealed AMA positivity associated with pruritus, the patient was referred to gastroenterology. The progress in clinical and laboratory values was taken as an indication of a decrease in liver inflammation as a liver biopsy was not performed. After UDCA treatment, both pruritus and renal function tests improved. On the other hand, it was predicted that the immune system modulated by this specific treatment could reduce the formation of immune complexes and their accumulation in the kidneys.9,10 According to the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, the indication for immunosuppressive treatment in primary MPGN is the presence of proteinuria at a nephrotic level. In this case, it is not a primary MPGN. With the treatment of the underlying pathology, the nephrological examinations began to improve, the serum creatinine level decreased, and the proteinuria decreased to 1,400 mg. In this case, it was decided to continue non-immunosuppressive supportive treatment. The patient was not treated with steroids or other immunosuppressive drugs in both the nephrology or gastroenterology department.
Therefore, the authors controlled secondary iMPGN associated with PBC with UDCA, which is used in the treatment of PBC, by prioritising liver inflammation. The authors knew that secondary MPGN can occur in the context of autoimmune diseases, but cases like this are very rare. Every patient with iMPGN should be investigated for secondary aetiologies.