
INTRODUCTION
In focal segmental glomerulosclerosis (FSGS), some (not all) glomeruli are affected with sclerosis (focal), and each diseased glomerulus is only partially affected (segmental).1-3 Sclerosis means renal vasculature is affected in a way similar to arteriosclerosis. Specifically, there occurs stiffness and obstruction in the vessels.2,3 Initially, juxtamedullary area is affected with sclerosis.3 As the disease advances, there is accumulation of glomerular collagen (Type IV).2 This proteinaceous material appears glassy pink on haematoxylin and eosin stain, and is called hyalinosis.3 FSGS is classified into primary FSGS, secondary FSGS, genetic FSGS, and FSGS of undetermined cause (FSGS-UC).4 If kidney biopsy reveals FSGS lesions, and patient has nephrotic syndrome (24 hours proteinuria >3.5 g and presence of serum albumin <30 g/L, with or without oedema), then there is more probability of primary FSGS.4 This is especially true if the electron microscopy shows diffuse foot process effacement.4 If the patient does not satisfy this criterion, then they should be investigated for secondary FSGS.4 Secondary FSGS could occur due to diabetic nephropathy, hypertensive nephrosclerosis, obesity, viral infections (including severe acute respiratory syndrome coronavirus 2, HIV, cytomegalovirus, Epstein–Barr virus, Hepatitis C virus, and Parvovirus B19), drug induced (including due to nonsteroidal anti-inflammatory drugs, steroids, calcineurin inhibitors, mammalian target of rapamycin inhibitors, and lithium), and reduced nephron number (as in sickle cell disease, age -related FSGS, renal dysplasia, and reflux nephropathy).1,4 Genetic testing to exclude genetic forms of FSGS (mutations in podocyte and glomerular basement membrane proteins) may be needed when subject does not meet the criteria for primary FSGS.4 In this paper, the authors present a histopathological discussion on FSGS.
DISCUSSION
Kidney Disease: Improving Global Outcomes (KDIGO) has recently recommended following clinical-pathologic or aetiologic classification of FSGS.4
Primary FSGS
KDIGO has recommended that when a patient has nephrotic syndrome, and in kidney biopsy, the light microscopy shows focal and segmental glomerulosclerosis lesions, and the electron microscopy shows diffuse foot process effacement, then it satisfies the recommended definition of primary FSGS.4 However, please note that no histopathological finding is pathognomonic of primary FSGS. Nephrotic syndrome is defined as 24-hour proteinuria of greater than 3.5 g, plus hypoalbuminaemia of less than 30 g/L, often with or without dyslipidaemia and oedema.4 There should not be any established pathophysiologic entity that may cause FSGS (no secondary factors that can cause FSGS should have been identified).4
Secondary FSGS
When a patient has FSGS lesions in presence of secondary factors that are known to cause FSGS, then it is termed as secondary FSGS. Electron microscopy may or may not show diffuse foot process effacement.4 Secondary causes of FSGS include, but are not limited to, diabetic nephropathy, hypertensive nephrosclerosis, obesity, viral infections (including severe acute respiratory syndrome coronavirus 2, HIV, and Hepatitis C), drug induced (including due to nonsteroidal anti-inflammatory drugs, steroids, calcineurin inhibitors, mammalian target of rapamycin inhibitors, heroin, interferon-α, and pamidronate), and reduced nephron number (as in age-related FSGS, renal dysplasia, surgical ablation, and reflux nephropathy).1,2,4
Genetic FSGS
Mutations in podocyte or glomerular basement membrane proteins may result in genetic FSGS.4 The mutations may occur in proteins including podocin, nephrin, α-actinin-4, and β-integrin.3 Patients with genetic forms of FSGS may have a family history of kidney disease, and they are often young.4
FSGS of Undetermined Cause
When no secondary cause of FSGS is identified, there is no identifiable genetic cause, nephrotic syndrome is not present, and electron microscopy does not reveal diffuse foot process effacement, then this type of FSGS is termed as FSGS-UC.4
HISTOPATHOLOGIC FINDINGS IN FSGS
Please refer to Figures 1–3 in this paper that showcase the FSGS histopathology.
Figure 1A shows three glomeruli. Among the three glomeruli, one of them shows segmental sclerosis and adhesion (periodic acid–Schiff [PAS] stain, x10 magnification). Figure 1B shows segmental sclerosis in glomerular tuft. The sclerosis involves less than 25% of the overall area. There is synechiae formation at the tip region (PAS, x40 magnification). Figure 1C shows sclerosis in glomerular tuft with obliteration of capillary lumen due to foamy histiocytes (periodic Schiff-methenamine silver, x40 magnification).
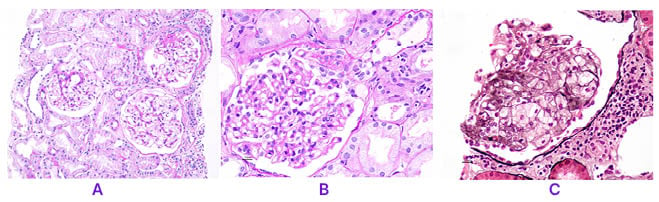
Figure 1: Focal segmental glomerulosclerosis histopathology (periodic acid–Schiff stain)
A) Among the three glomeruli, one of them shows segmental sclerosis and adhesion (PAS, x10).
B) Glomerular tuft shows segmental sclerosis involving less than 25% of the overall area, and synechiae formation at the tip region (PAS, x40).
C) Glomerular tuft shows sclerosis with obliteration of capillary lumen due to foamy histiocytes (PASM, x40).
PAS: periodic acid–Schiff stain; PASM: periodic Schiff-methenamine silver.
Figure 2A shows glomerulus with reactive and hyperplastic podocytes with collapse of underlying capillary tufts (Masson’s trichrome, x40 magnification]. Figure 2B shows segmental hyalinosis/sclerosis in the perihilar region. Masson’s trichrome stains bluish color in the solidified area (x40 magnification).
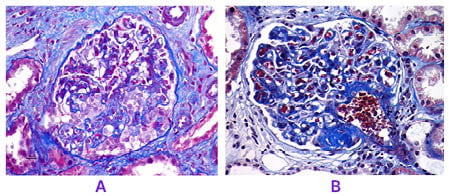
Figure 2: Focal segmental glomerulosclerosis histopathology (Masson’s trichrome stain)
A. Glomerulus revealing reactive and hyperplastic podocytes with collapse of underlying capillary tufts (MT, x40).
B. Segmental hyalinosis/sclerosis is evident in the perihilar region. MT stains bluish color in the solidified area (MT, x40).
MT: Masson’s trichrome.
Figure 3 shows glomerular tufts with negative staining with a panel of antisera. This image is with IgG antisera (x40 magnification).
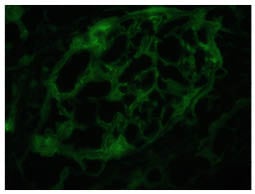
Figure 3: Glomerular tufts reveals negative staining with panel of antisera.
This image is with IgG antisera (x40).
Light Microscopy
• Focal glomerulosclerosis (only some glomeruli are affected) and segmental glomerulosclerosis (only a part of a glomerulus is affected). The sclerosing segments are positive with PAS and silver-methenamine stains that stain Type IV collagen.1,2
• As immunologic mechanism is not fundamental to pathogenesis of FSGS. Cell proliferation is not a characteristic feature in FSGS. Mesangial cell proliferation was believed to be a feature of FSGS in the past, and could be confused with other diseases, including IgA nephropathy coexisting with FSGS.1-3
• Mesangial sclerosis that appears to begin in corticomedullary region.1-3
• Hyaline deposits below glomerular basement membrane.1,2
• Endocapillary foam cells or lipoid droplets.1,2
• Tubular atrophy, interstitial fibrosis, and hyaline thickening of afferent arterioles (focal lesions).1,2
Immunofluorescence
Because FSGS is non-immunologic in pathogenicity, immune deposits are not a characteristic feature. However, the sclerotic areas may show focal and segmental deposits of IgM and C3, particularly where hyaline material is deposited.1,3
Electron Microscopy
• Detachment of podocytes from glomerular capillary basement membrane. The space thus created between podocytes and the basement membrane is filled with collagen, giving rise to an appearance of a halo, like halo of the moon. This halo stains paler blue (when compared with the vessels) in trichrome stain.1,3
• Extensive foot process effacement, even in non-sclerotic glomeruli.1,3 In primary FSGS, podocyte foot process effacement is more severe, typically involving >80% of the basement membrane’s surface area. The effacement in secondary FSGS is more uneven and patchy.5
• Wrinkled basement membrane.1,3
• Foamy macrophages.1,3
• Intracytoplasmic vacuoles may form in podocytes.1,3
• Proliferation of podocytes may occur.1-3
• Mesangial sclerosis with increased mesangial matrix.1,3
• Collapsed glomerular loops.1,3
Please note that sometimes in patients with FSGS, needle biopsy may sample a renal tissue that does not have FSGS lesions. In that case, FSGS diagnosis may be missed. The lesions picked up may instead suggest minimal change in disease.
HISTOLOGIC CLASSIFICATION OF FSGS, OR COLUMBIA CLASSIFICATION
FSGS Not Otherwise Specified
This is the most common form. This is a diagnosis of exclusion; this means the other variants (perihilar, cellular, tip, and collapsing) must be excluded before coining the term FSGS not otherwise specified to these lesions.2,3 In thisvariant, we see focal and segmental lesions with increased extracellular matrix, and obliteration of the glomerular capillary lumen.2,3
FSGS Perihilar Variant
Before diagnosing FSGS lesions as perihilar variant, one must exclude hypercellular and collapsing variants. If there are some perihilar variant lesions, but even one hypercellular or collapsing variant lesion, then this is not called a perihilar variant, but a hypercellular or collapsing variant, respectively.2 Presence of tip variant does not exclude perihilar variant. Perihilar variant is more commonly seen in secondary FSGS.3 The requirements to diagnose perihilar variant are:
• There should be at least one glomerulus with perihilar hyalinosis, with or without sclerosis;2,3 and
• Perihilar sclerosis and/or hyalinosis must be present in more than 50% of the glomeruli with segmental lesions.2,3
FSGS Cellular Variant
The tip and the collapsing variants must be excluded before the diagnosis of FSGS as cellular variant.2To diagnose this cellular variant, there must be at least one glomerulus with endocapillary hypercellularity, including endothelial cells, macrophages, and foam cells.2 Foam cells are vascular endothelial cells or monocytes, and are laden with lipids.3 This endocapillary hypercellularity must involve at least 25% of the tuft and cause occlusion of the capillary lumen.2
FSGS Tip Variant
The tip refers to zone of the glomerular tuft adjacent to the proximal tubule.2,3 To diagnose the tip variant, one must exclude the collapsing variant.2,3 This means that even if one glomerulus has a collapsing variant lesion, one cannot diagnose this FSGS as tip variant.2 To call FSGS tip variant, at least one segmental lesion involving the tip domain must be present, with one of the following findings at the tubular lumen or neck:2
• Adhesion between Bowman’s capsule and the tuft.2
• Confluence of podocytes with parietal or tubular epithelial cells.2
FSGS tip variant has a favorable prognosis when compared with other variants.3
FSGS Collapsing Variant or Collapsing Glomerulopathy
If there is any lesion with characteristics of collapsing variant, the diagnosis of all other variants is excluded. In the collapsing variant, there must be at least one glomerulus with collapse and hypertrophy and hyperplasia of overlying podocyte.2 There may be retraction and collapse of the capillary walls.3 Hyperplasia of podocytes is associated with this variant.2,3 Severe proliferation of podocytes may be confused with crescent formation.3
SIGNIFICANCE OF THE PATHOLOGY FINDINGS IN TERMS OF THE RECOMMENDED TREATMENT
FSGS-UC and Secondary FSGS: KDIGO 2021 guidelines recommend that immunosuppressive medications should not be used to treat FSGS-UC and secondary FSGS, as there is lack of evidence to suggest any benefit of immunosuppression in this subclass of patients. Since risks outweigh benefits, the use of immunosuppressive medications in these patients should better be avoided.1 General measures to reduce proteinuria include medications for renin–angiotensin system blockade (including, but not limited to, angiotensin-converting enzyme inhibitors and angiotensin receptor blockers), optimal control of blood pressure, salt restriction, normalising weight, stopping smoking, and exercising regularly.1
Treatment of primary medical condition responsible for secondary FSGS is recommended in KDIGO 2021 guidelines.1
Primary FSGS
KDIGO guidelines recommend high-dose oral glucocorticoids as first-line immunosuppressive therapy in primary FSGS.1 Cyclosporin or tacrolimus are recommended in steroid-resistant primary FSGS.1
Histopathologic classification of FSGS helps in prognostic terms.5 The collapsing variant has worse prognosis, with approximately 70% of patients with this variant progressing to Stage 5 chronic kidney disease.5 Such progression in tip lesion, FSGS not otherwise specified (classic), perihilar, and cellular variants is less common, with 5–20%, 30–40%, 30–50%, and around 30% of patients progressing to Stage 5 chronic kidney disease, respectively.5
KEY LEARNING POINTS
1. Light Microscopy: in FSGS, there is focal glomerulosclerosis (only some glomeruli are affected) and segmental glomerulosclerosis (only a part of a glomerulus is affected). The sclerosing segments are positive with PAS and silver-methenamine stains that stain Type IV collagen.1,2
2. Immunofluorescence: FSGS is non-immunologic in pathogenicity. Immune deposits are not a characteristic feature. However, the sclerotic areas may show focal and segmental deposits of IgM and C3, particularly where hyaline material is deposited.1,3
3. Electron microscopy is an important tool to differentiate primary and secondary FSGS. Podocyte foot process effacement is more severe in primary FSGS, generally involving >80% of the basement membrane’s surface area. Secondary FSGS effacement is more uneven and patchy.5






