Abstract
Monoclonal antibodies targeting the epidermal growth factor receptor (EGFRI), such as cetuximab and panitumumab, are commonly used systemic therapies for advanced colorectal and head and neck cancers. Hypomagnesaemia is a common side effect of these therapies and occurs in up to 30% of patients. Interruption of EGFR signalling in the distal convoluted tubule leads to inactivation of the transcellular transporter transient receptor potential channel melastatin member 6 and increased renal magnesium excretion. This paper describes the incidence, risk factors, and the emerging management options for EGFRI-induced hypomagnesaemia.
INTRODUCTION
Over the past few decades, there have been important advances in cancer therapeutics, including a shift from traditional cytotoxic chemotherapy to targeting specific intra-cellular pathways and harnessing the immune system through immunotherapies. One important target is the epidermal growth factor receptor (EGFR), which is a member of the ErbB receptor kinase family. Activation of EGFR can occur through amplification, point mutations, or ligand excess and leads to a signalling cascade through mitogen-activated protein kinase, with downstream effects on the Ras/Raf/MEK/ERK and PI3K-AKT- (PTEN)-mTOR pathways. This overstimulation results in tumour invasion, growth, and metastasis and has been shown to be of oncogenic importance in glioblastomas, lung cancer, head and neck cancer, and colorectal cancer (CRC).1-3
Approximately 15% of non-small cell lung cancers harbour mutations in EGFR tyrosine kinase and tyrosine kinase inhibitors such as erlotinib, gefitinib, afatinib, and osimertinib have significantly improved survival for patients with EGFR-mutated advanced non-small cell lung cancers with life expectancies measured in years.4 In CRC and head and neck cancers, monoclonal antibodies that block ligand binding to EGFR, such as cetuximab (CTX) and panitumumab (PMAB), have been shown to improve survival in advanced disease.5,6
Although targeting the same pathway and sharing a similar toxicity profile (diarrhoea, rash, paronychia), EGFR tyrosine kinase inhibitors have rarely been associated with hypomagnesaemia (hMG), in contrast to EGFR inhibitors (EGFRIs), which may lead to hMG in up to 30% of patients. Often referred to as the ‘forgotten ion’, magnesium (MG) plays a pivotal role in many bodily processes through its role as a cofactor for many enzymatic reactions and serving as a structural component of proteins and nucleic acids. Manifestations of hMG can be quite variable, ranging from asymptomatic to non-specific symptoms such as fatigue and nausea. Lethal manifestations may include arrhythmias, seizures, and tetany.7
In this review, the authors will explore the mechanism, risk factors, and current management strategies for hMG induced by EGFRIs.
EPIDERMAL GROWTH FACTOR RECEPTOR INHIBITORS IN ONCOLOGY
The original Phase I studies of EGFRIs investigated CTX as a single agent or in combination with cisplatin in patients with EGFR expressing tumours (mainly head and neck and lung cancers).8 CTX is a chimeric IgG1 monoclonal antibody targeting the EGFR pathway by competitively inhibiting the extracellular domain of the EGFR, as well as leading to dimerisation and downregulation of EGFR.
In advanced CRCs (ACRC), expression or upregulation of the EGFR gene is present in 60–80% of tumours, making EGFR an attractive target.6 In patients with EGFR expressing irinotecan refractory metastatic colon cancer, the addition of CTX to irinotecan resulted in improved response rates and progression-free survival (PFS) compared with CTX monotherapy alone.6,4 Acne-like rash occurred in 80% of patients on this trial; however, since MG was not collected as part of the study procedures, information about the frequency of hMG was not reported. In the landmark National Cancer Institute (NCI) of Canada trial CO.17, CTX was compared with best supportive care (BSC) in patients with EGFR expressing refractory ACRC and showed an improved overall survival (OS) benefit.9 A follow-up biomarker analysis of mutation of the KRAS gene identified KRAS as an important predictive biomarker, as those with a gene mutation did not derive benefit from CTX, while those with wild-type KRAS had significant OS benefit from the EGFRI (hazard ratio: 0.55; 95% confidence interval [CI]: 0.41–0.74).10 The CO.17 trial also collected MG as part of the study procedures and found that 53% of those treated with CTX developed hMG versus 15% on BSC.
PMAB is a humanised IgG2 monoclonal antibody against EGFR and has also been found to improve response rates and PFS in KRAS wild-type tumours compared to BSC in refractory ACRC.11 In this trial, hMG was observed in 36% of patients receiving PMAB compared with 1% in the BSC arm.12 Interestingly, in the non-inferiority trial of PMAB compared with CTX in ACRC, any grade hMG (27.6% versus 18.1%) and Grade 3/4 hMG (7.0% versus 2.8%) was higher with the use of PMAB.13
Advances in the knowledge of the predictive impact of RAS and primary tumour location in the selection of initial therapy for untreated ACRC has led to EGFRIs (PMAB or CTX) being combined with a chemotherapy backbone (FOLFOX/CAPOX or FOLFIRI) in patients with RAS/BRAF wild-type and left-sided primary tumours.14,15 These combinations have resulted in a median OS approaching 3 years in this population, but have also been associated with high rates of any grade hMG (>63%), including Grade 3/4 hMG (4–8%).16-18
EGFRIs have also played a role in the treatment of head and neck cancer, where the combination of CTX and radiation for localised disease was shown to improve survival when compared with radiation alone.19 In the recurrent or metastatic disease setting, CTX and platinum plus 5-fluorouracil resulted in improved OS compared with chemotherapy alone.5 The incidence of Grade 3/4 hMG in CTX plus chemotherapy arm was significantly greater than the chemotherapy alone arm, occurring in 5% and 1% of cases, respectively. Multiple other studies have established the role of EGFRIs in the world of oncology.
PHYSIOLOGY OF MAGNESIUM HOMEOSTASIS
MG is the fourth most commonly abundant cation in the body and is the second most common intracellular cation after potassium.20 It is the cofactor for over 300 enzymatic reactions and is involved in stabilising enzymes including many adenosine triphosphate-generating reactions. It plays an essential role in cellular processes, as it is involved in important physiologic functions such as nucleic acid metabolism, protein synthesis, and energy production.21,22 MG is also crucial for muscle contraction and relaxation, nerve function, heart rhythm, vascular tone, and bone formation.20,21
Distribution, Absorption, and Excretion
MG is located intracellularly in 99% percent of the total body, which includes bones, skeletal muscles, and non-muscular soft tissues, leaving only 1% in the extracellular space (serum and red blood cells).21 The kidneys easily filter 70% of the total body MG, either as complex anions (oxalate, phosphate, citrate) or ionised MG.21,23 The remaining 30% of the total body plasma MG is bound to proteins, mainly albumin.21,23
MG’s homeostasis is mainly regulated by the intestines, kidneys, and bones. MG is absorbed in the gut and stored in bones, and the excess is excreted by the kidneys and intestines.23 The majority of MG (90%) is absorbed in the ileum, with some absorption in the colon via a paracellular mechanism, which is driven by an electrochemical gradient and solvent drag.20 The second transport system for MG occurs in the cecum and colon, using the transcellular transporter transient receptor potential channel melastatin member (TRPM) 6 and TRPM7.20,23 The latter is an active process and accounts for about 10% of MG reabsorption.23 About 30–50% of the dietary MG intake is absorbed in the gut. The amount of MG absorbed is related to the MG status: the lower the MG level, the more the mineral will be absorbed.21
The Kidney’s Role in Handling Magnesium
The kidneys play an important role in MG homeostasis, as non-protein bound MG is freely filtered across the glomerulus to maintain proper serum MG concentration.20,21 MG excretion follows the circadian rhythm as the majority of excretion occurs at night.20 Under physiological conditions, 95% of filtered MG is reabsorbed by the kidneys and 3–5% is excreted in the urine.20,21 Reabsorption sites include the proximal tube and more commonly (60–70%) the thick ascending limb of the loop of Henle via a passive paracellular transport process.20 A small percentage (10%) is reabsorbed at the distal convoluted tubules (DCT) for the fine-tuning of MG excretion via an active transcellular transport process.20 The entry of MG into the DCT cells is facilitated by TRPM6.22 The mechanism of activation of this transport channel has been linked to binding of EGF to the EGFR on the DCT cells.24 EGFRIs bind to EGFR and inhibit the influence of EGF, which leads to decreased activation of the TRPM6 channels and a decrease in MG reabsorption in the body24 (Figure 1).
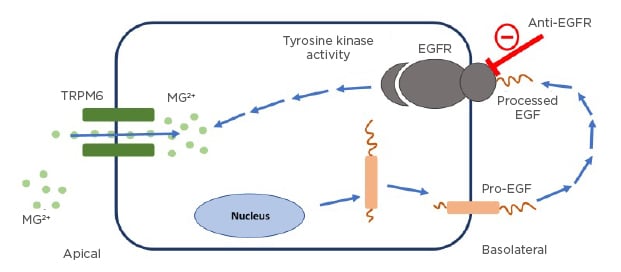
Figure 1: The role of the epidermal growth factor receptor pathway interruption in causing hypomagnesaemia.
Pro-EGF mutation leads to impaired stimulation of EGFR, as shown above. EGFR activation is necessary for TRPM6 channel activation to prevent renal magnesium wasting. EGFRIs work by interrupting this pathway resulting in renal MG wasting.
EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; MG: magnesium; TRPM6: transcellular transporter transient receptor potential channel melastatin member 6.
Adapted from Costa et al.23
ASSESSMENT OF MAGNESIUM STATUS
Several methods are utilised to measure MG levels; however, the most common method used is testing the serum MG concentration.21 Unfortunately, this is a poor predictor of the MG concentration in the body, as serum MG only accounts for 0.3% of the total body MG.21 In oncology, the degree of hMG is assessed using the Common Terminology Criteria for Adverse Events (CTCAE) v4.03, with Grade 1 defined as <lower limit normal–0.5 mmol/L (1.2 mg/dL); Grade 2 as <0.5–0.4 mmol/L (<1.2–0.9 mg/dL); Grade 3 as <0.4–0.3 mmol/L (<0.9–0.7 mg/dL); and Grade 4 as <0.3 mmol/L (<0.7 mg/dL Table 1).25 Other than EGFRIs, which lead to hMG by reducing kidney reabsorption, other causes of hMG include poor dietary intake, gastrointestinal losses, kidney losses, and endocrine diseases (Table 2).20,21 It is important to assess for hMG in patients on EGFRIs as symptoms are often non-specific and may overlap with symptoms of cancer therapy and/or cancer progression. Clinical manifestations include neuromuscular signs (tremor, spasticity, weakness, ataxia, tetany, and cramps), cardiovascular symptoms (prolonged QT interval and ventricular arrhythmia), and neurocognitive dysfunction (depression, cognitive impairment, agitation, psychosis, and seizures).20,21,26 In addition, MG is involved in the regulation of the parathyroid hormone and, therefore, hMG can lead to hypocalcaemia (hCA).26
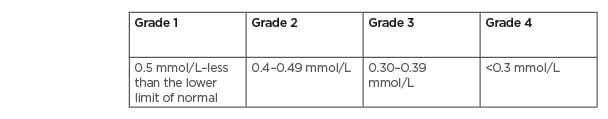
Table 1: Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 hypomagnesaemia grading.2
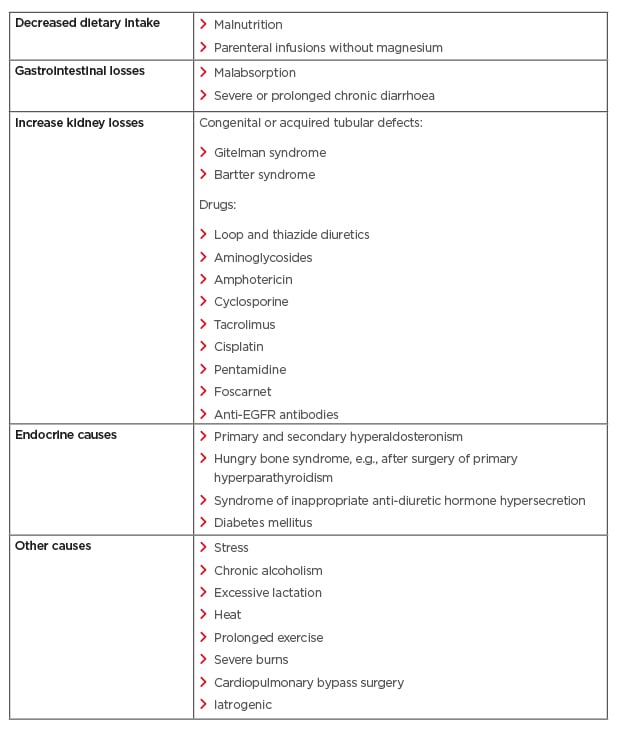
Table 2: Various aetiologies of hypomagnesaemia.
CHARACTERISTICS OF EPIDERMAL GROWTH FACTOR RECEPTOR INHIBITOR-INDUCED HYPOMAGNESAEMIA
The first report of EGFRI-induced hMG was in a 34-year-old male patient with metastatic CRC receiving irinotecan and CTX, where symptoms manifested as profound fatigue, paraesthesia, muscular fasciculations, and symptomatic hCA.27 Investigations revealed inappropriately high levels of urinary MG consistent with renal wasting. The patient’s symptoms resolved with intravenous (IV) repletion only to recur within 48 hours of oral supplementation with MG oxide and calcium carbonate. Ultimately, the patient required nightly infusions of 10 g MG sulfate to maintain his MG levels and allow ongoing CTX based therapy.
A further prospective study by Tejpar et al.28 evaluated 98 patients with CRC being treated with CTX, with or without chemotherapy, and used 16 patients on treatment with chemotherapy alone as a control group. This study showed that 97% of patients treated with CTX developed progressive hMG. The mean serum MG slope during EGFRI treatment (with or without combined chemotherapy) was significantly lower compared with chemotherapy alone (-0.00157 mmol/L/day; standard deviation: 0.00162 [95% CI: -0.00191−-0.00123] versus 0.00014 mmol/L/day; standard deviation: −00076 [95% CI: -0.00026–0.00055]; t-test p<0.0001). The degree of hMG was correlated with the duration of treatment and patients treated for under 12 weeks who did not develop Grade 3/4 hMG. Higher baseline serum MG concentration and increasing age were identified as factors that inversely correlated with hMG development. An IV MG load test (N=5) confirmed a defect in renal MG reabsorption as the underlying mechanism of hMG, and nicely illustrated why intermittent boluses of IV MG are an ineffective strategy for the management of EGFRI-induced hMG.
Similarly, retrospective studies have showed direct relationships between older age and duration of EGFRI therapy on risk of developing hMG.29,30 A systematic review by Jiang et al.26 also found that length of EGFRI treatment, concomitant platinum chemotherapy, increasing age, and baseline MG concentration correlated with severity of hMG. The effects of EGFRIs on MG typically resolve within weeks to a few months after discontinuation of EGFRI therapy.26,31 In addition, a meta-analysis of prospective clinical trials involving CTX and PMAB revealed all-grade incidence to be 34.0%, 14.5%, and 16.8% for the development of hMG, hypokalaemia, and hCA, respectively.32 PMAB had a similar incidence of all grade hMG to CTX (31.8% versus 34.9%), but possibly a higher risk of Grade 3/4 hMG (5.4% versus 4.4%). For CRC specifically, Grade 3/4 hMG was higher for PMAB at 4.6% versus 2.9% for CTX. Likewise, the ASPECCT trial, which compared the efficacy of CTX with PMAB in chemotherapy-refractory ACRC, found that PMAB was associated with more severe hMG (Grade 3/4: 7.0% versus 2.8%).13
Since off-target EGFRI side effects such as acneiform rash have been associated with improved response rates and survival,9,33 there has been interest in EGFRI-induced hMG as a surrogate predictive biomarker for improved outcomes. In an initial study involving 68 patients treated with CTX plus irinotecan, a 20% (and subsequently 50%) MG reduction from baseline was associated with improved response rates, time to progression, and OS.34,35 In contrast, an analysis of the CO.17 clinical trial (CTX versus BSC) found that Grade ³1 and ³20% reduction in hMG from baseline was associated with worse OS.36 Subsequent secondary analyses and meta-analyses have suggested that EGFRI-induced hMG is associated with improved clinical benefits (PFS and OS).13,37 It is important to note that studies showing a positive correlation with worse hMG and improved survival are subject to bias, since patients have a higher chance of developing hMG if they have disease control (and longer survival) on EGFRIs because they are receiving EGFRIs for a greater duration, in contrast to those that have early disease progression and cease EGFRIs earlier.38 This guaranteed time bias (also known as immortal time bias) can be overcome by using a landmark approach as was used in the CO.17 hMG analysis.36 As a result, it is unclear at present if the development of hMG can be used to predict for improved outcomes with EGRIs.
MANAGEMENT OF EPIDERMAL GROWTH FACTOR RECEPTOR-INDUCED HYPOMAGNESAEMIA
While hMG is generally easily correctable, when it is EGFRI-induced, it can be very challenging to manage. This is a result of ongoing renal losses due to EGFRI effects on the TRPM6 MG channel in the DCT and also due to impaired transcellular intestinal MG absorption from similar inhibition of TRPM6 in the gut.24,39 These effects on absorption and excretion make oral and IV supplementation problematic.
Early institutional practices favoured 4 g of IV MG sulfate starting at Grade 2 hMG and increasing to 6–10 g IV MG daily or twice weekly for Grade 3/4; however, it was noted that neither IV or oral magnesium replacement sustained magnesium repletion beyond 72 hours.20,40 Oral MG supplementation may cause diarrhoea, while frequent IV administrations are very inconvenient and time consuming for patients, and may exacerbate renal MG leak as a result of altered TRPM6 signalling.28,40,41 Recent opinion-based guidelines suggest consideration of oral MG for Grade 1/2 and adding regular IV MG infusions at Grade 2/3 hMG.42 With the development of Grade 4 hMG, discontinuation of EGFRIs is suggested until MG improves to Grade 2 or less.
A survey of Canadian medical oncologists found that most physicians (>90%) regularly monitor MG prior to each EGFRI infusion, and most employ a reactive MG replacement strategy (as opposed to prophylactic).43 Forty percent of respondents (N=40) favoured IV supplementation alone, while 45% used both oral and IV, and 70% introduced supplementation at Grade 1 hMG and the remainder at Grade 2. Importantly, 30% of oncologists were withholding EGFRIs at Grade 3 hMG and 45% at Grade 4. The vast majority felt that a consensus guideline on the management of EGFRI-induced hMG was necessary given the confusion about the significance of this side-effect and the best replacement strategy. In an attempt to explore the availability of evidence-based management practices for EGFRI-induced hMG, Jiang et al.26 performed a systematic review and found a lack of high-quality management strategies and the available reports suggested refractoriness to IV and oral replacement. As a result, prospective comparative trials were recommended.
EGFRIs inihibition of EGFR and resultant downstream effects on the TRPM6 channel in the kidney and intestine is analagous to an inherited syndrome called familial hMG with secondary hCA. This rare autosomal, recessive condition is due to a defect in the TRMP6 gene on chromosome 9q22, which encodes the magnesium-permeable ion channel.44 Patients with this condition have severe hMG and hCA as neonates, which can lead to intractable seizures, cerebral damage, and death. If recognised early, patients may be spared morbidity as long as they are maintained on a high dose of oral MG supplementation. Other evidence supporting an oral supplementation approach to EGFRI-induced hMG comes from Pietropaolo et al.39 and studies on the effects of CTX on intestinal MG absorption. They showed that CTX does indeed affect active transcellular transport through intestinal TRPM6 and, that by increasing oral MG supplementation and thus intestinal MG concentrations, paracellular transport may be the most effective strategy to combat EGFRI-induced hMG.
Recently, the results of the randomised multicentre MAGNET trial became available, which focused on early oral MG supplementation.45 Eighty-nine patients were randomised to a reactive strategy of oral MG gluconate 3 g twice daily (BID) at the occurrence of Grade 1 hMG, while 84 patients were randomised to a prophylactic strategy of oral MG gluconate 3 g BID at initiation of EGFRI and increasing to 6 g six times a day at Grade 1 hMG. The slope of decline of MG was significantly steeper with the reactive strategy, and 13% developed hMG in the reactive arm compared with only 4% in the prophylactic arm. Importantly, oral supplementation was well tolerated, with no significant adverse events and no difference in bowel movements between the two arms. This study suggests that a prophylactic oral MG strategy may be effective in preventing significant hMG. Another prospective trial46 assessed the feasibility of using reactive oral MG replacement strategies, initiated at the initial development of Grade 1–3 hMG for patients receiving platinum-based chemotherapy or EGFRIs. Patients were randomised 1:1 to MG oxide 420 mg per os BID or MG citrate 150mg per os BID (and titrated up depending on the grading of hMG). The trial failed to accrue sufficient numbers, but of the 15 randomised patients MG levels stabilised with positive slopes of change in MG from the baseline. Few patients required IV MG replacement and only 20% developed Grade 1 diarrhoea (Vickers, personal communication, 2021).
Given the increasing use of EGFRIs in earlier lines of therapy for ACRC, managing hMG will be a common challenge for oncologists. In combination with cytotoxic chemotherapy, EGFRI-induced hMG may occur in over 50% of patients and up to 30% on monotherapy EGFRIs. Routine monitoring of MG levels is required, and recognition of risk factors such as advancing age, longer duration of EGFRI therapy, and concomitant platinum chemotherapy is important.26 In addition, comorbidities such as diabetes, renal tubular disorders, hyperthyroidism, hyperaldosteronism, refeeding syndrome, and the postoperative setting should be taken into account.21,26 Physicians should also consider electrocardiograms for baseline QTc assessments, as hMG could exacerbate the risk of cardiac arrhythmias. From a management perspective, physiologic studies and prospective trials support the use prophylactic oral MG supplementation at initiation of EGFRIs, or early after hMG develops.39,45 These studies also show that oral supplements appear to be well tolerated and the requirement for IV MG may be avoided or delayed until more severe hMG develops. Due to the variable availability of MG supplements across the world, a specific MG supplement cannot be universally recommended; however, a diet high in MG and upward titration of oral MG supplementation is suggested to saturate intestinal MG concentrations for paracellular absorption. IV MG supplementation should mainly be administered in patients requiring immediate correction such as for ventricular arrhythmias, or those at risk of other complications related to moderate or severe hMG.20 Future trials focusing on efficacy, tolerability, and quality of life of patients receiving different oral MG supplements are required.
CONCLUSION
hMG is a common side effect of treatment with EGFRIs, with some patients more susceptible to EGFRI-induced hMG than others. Recent investigations have provided insights that support the use of early oral MG supplementation. Clinicians should closely monitor for this side effect and consider early implementation of an oral MG replacement to avoid serious complications and achieve the best therapeutic results from EGFRIs.