Non-steroidal anti-inflammatory drugs (NSAID) are one of the most popular analgesics due to their efficacy and availability over the counter.1 However, there are increasing safety concerns about NSAID side effects.2 Inhibition of prostaglandin synthesis within the kidney, which impairs renal perfusion, is the main mechanism of NSAID-induced nephrotoxicity.3 Diclofenac, a prominent NSAID, is claimed to evoke kidney damage even in patients without previous renal dysfunction.4 Experimental data suggest that the mechanism of diclofenac’s action may go beyond cyclo-oxygenases inhibition in a way that is responsible for the drug’s toxicity.5
Overactive glutamate signalling in the kidney leads to renal pathologies, and as such glutamate receptor antagonists are claimed to be nephroprotective;6 however, results of other studies remain controversial.7 Kynurenic acid (KYNA), a metabolite of tryptophan, is synthesised from L-kynurenine by kynurenine aminotransferases (KAT).8 KAT I and KAT II isoenzymes are the most studied KAT isoforms.9 The main mechanism of KYNA action is nonselective antagonism towards ionotropic glutamatergic receptors, especially N-methyl-D-aspartate types, which are predominantly expressed in the kidney. Natriuretic, anti-inflammatory, and hypotensive properties of KYNA are well established.10
The aim of this study was to examine the effect of diclofenac, one of the strongest and most commonly prescribed NSAID, on KYNA formation and the activity of KAT I and KAT II, in rat kidney in vitro. Furthermore, the molecular docking of diclofenac to KAT I and KAT II structures was conducted to study the mechanism of drug-enzyme interaction. This was followed by microarray data mining to investigate whether diclofenac can affect the expression of KAT-coding genes.
Diclofenac at 500 µM and 1 mM lowered KYNA formation in kidney homogenates in vitro to 67% (p<0.001) and 36% (p<0.001) of control value, respectively. At 500 µM and 1 mM concentration, diclofenac decreased renal KAT I activity in vitro to 40% (p<0.05) and 41% (p<0.01) of control value, respectively. Additionally, diclofenac at 50 µM, 100 µM, 500 µM, and 1 mM concentration decreased kidney KAT II activity in vitro to 88% (p<0.01), 62% (p<0.01), 11% (p<0.01), and 4% (p<0.01) of control value, respectively. Molecular docking results suggested that diclofenac may interact with the active site of KAT I and KAT II. Publicly available microarray datasets suggested that diclofenac does not affect the expression of KAT-coding genes.
To summarise, diclofenac decreases KYNA production in rat kidney in vitro by inhibiting KAT I and KAT II isoenzymes. Results of this study show a novel mechanism of diclofenac action in the kidney. Its relation to drug induced nephrotoxicity should be clarified in future studies.
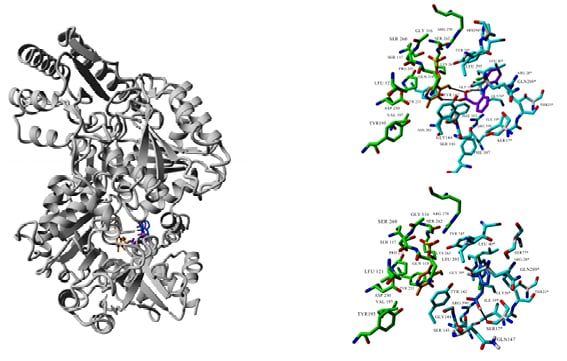
Figure 1: Molecular docking of diclofenac (two orientations: purple and blue) to the crystal structure of KAT II (PDB ID: 2R2N). Ligand and co-factor PMP (orange) are rendered in stick mode, residues involved in ligand and PMP binding are shown in cyan and green, respectively.
KAT II: kynurenine aminotransferase II; PMP: pentamannosyl 6-phosphate.