Abstract
Congenital heart disease represents the most prevalent birth defect, and surgical and interventional advances have translated to a burgeoning adult population of patients. Timely, tailored, and lesion-specific interventions are necessary to optimise long-term outcomes for this complex, heterogeneous patient cohort. We present a review of the most common defects which may be encountered in general adult cardiology. Particular focus is paid to the recommended interventional options and associated complications for each condition in line with European and American guidelines.
INTRODUCTION
Congenital heart defects are the most frequently occurring birth anomaly, affecting around 8 in every 1,000 live births.1 This corresponds to 1.35 million live births with congenital heart disease (CHD) worldwide annually,2 representing a significant global health burden. With advances in cardiac surgery and perioperative and medical care, over 85% of children with CHD survive into adulthood.3 This has expanded the population of adults with CHD to an estimated 13 million worldwide.4 Excluding spontaneously healed defects and bicuspid aortic valve disease, the prevalence of adult CHD is 3.5 per 1,000 adults.5
Since Alfred Blalock and Helen Taussig6 pioneered the first subclavian to pulmonary artery (PA) shunt, which “turned a blue baby pink” in 1944, congenital heart interventions have been developed for even the most complex lesions. The advent and application of cardiopulmonary bypass for CHD surgery was a major milestone in the mid-1950s. Catheter interventions in CHD began with William Rashkind and William Miller’s7 development in 1966 of balloon atrial septostomy for palliation of neonates with transposition of the great arteries. Thereafter, since the 1980s, there has been an explosion in the number and breadth of minimally invasive catheter-driven procedures, which has transformed the field of CHD.
Advances in surgical techniques and improvements in perioperative care have reduced surgical mortality to the single digits for most operations. Technological advances in imaging techniques have facilitated even antenatal diagnoses and therapies. Guidelines summarising and evaluating recent evidence have been developed to guide management strategies for individual conditions, including the timing and indications for repair and follow-up.8,9 However, the heterogeneity of disease processes and small patient numbers along with the speciality’s relative youth, has meant a lack of robust evidence. All of the recommendations stated in this review are based on expert consensus (Level C evidence), unless stated otherwise.
This review aims to introduce the non-specialist to repair of the five most common congenital heart defects seen in adults: atrial septal defect (ASD), ventricular septal defect (VSD), patent ductus arteriosus (PDA), coarctation of the aorta (CoA) and pulmonary stenosis (PS). Tackling the more complex lesions is beyond the scope of this manuscript.
ATRIAL SEPTAL DEFECT
ASD is the second most common type of CHD worldwide,10 affecting 1.3 per 1,000 live births,11 and, as such, is frequently encountered by the general cardiologist. The term encompasses four distinct structural entities, with only the first two affecting the true atrial septum: secundum ASD (80% of ASDs), primum ASD (15%), sinus venosus defect (5%), and unroofed coronary sinus (<1%). ASDs often present incidentally in children, but advancing age harbours increasing rates of exercise intolerance, atrial tachyarrhythmias, right heart enlargement and dysfunction, and pulmonary hypertension (PH). Consequently, adults with uncorrected defects have shortened life expectancies.12
Internationally recognised indications for ASD closure are summarised in Table 1a.8,9 Patients with ASDs not fulfilling these criteria should have ongoing follow-up, as shunt size can increase with age.13 Pulmonary vascular resistance >8 WU or shunt reversal with Eisenmenger physiology are contra-indications to closure (Class III).
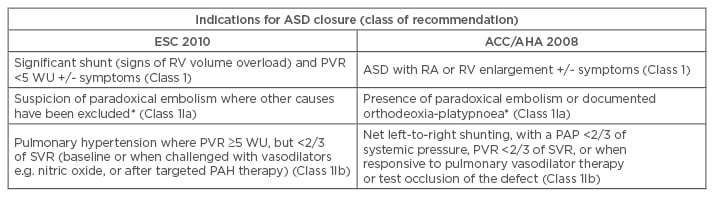
Table 1a: Indications for intervention in ASD.
*This is an indication for intervention in an ASD of any size.
PAP: pulmonary artery pressure; ESC: European Society of Cardiology; ACC/AHA: American College of Cardiology/American Heart Association; PAH: pulmonary arterial hypertension; PVR: pulmonary vascular resistance; Qp/Qs: pulmonary flow/systemic flow; RA: right atrium; RV: right ventricle; SVR: systemic vascular resistance; WU: wood units; ASD: atrial septal defect.
Adapted from the 2010 ESC guidelines8 and the 2008 ACC/AHA guidelines.9
ASD closure may take the form of surgical repair or transcatheter device closure. Percutaneous closure is first-line in secundum ASDs with a stretched diameter <36–40 mm and a sufficient rim of tissue to anchor the device. In this population, catheter intervention has a successful closure rate of >98% with an acceptable procedural safety profile. The major complication rate is quoted at 1.6%14 and includes erosion into surrounding structures, device thrombosis with systemic thrombo-embolism, device embolisation requiring surgery, pericardial tamponade, and death. Minor complications (1–5%) include atrial tachyarrhythmias, transient heart block, and vascular complications. Delayed complications following percutaneous closure include device endocarditis, nickel hypersensitivity, migraine, and conduction abnormalities.15 Aspirin is recommended for 6 months following device closure, with institution specific decisions regarding extended antiplatelet therapy beyond this period.16
Surgical closure is required in ASDs (both secundum and non-secundum) where the anatomy precludes device closure. Modern surgical techniques involve open suture or patch closure on cardiopulmonary bypass or minimally invasive, video-assisted thoracoscopic surgery. Operative mortality is low (<1%), although advancing age and comorbidities increase morbidity, including congestive cardiac failure and longer intensive therapy unit stays.12
Repair normalises life expectancy when performed before the age of 25 years, and may improve exercise tolerance and right heart function when performed at any age.17,18 Very long-term follow-up continues to provide insights into the effects of early repair, with a single-centre study of surgical ASD closure patients over four decades reporting right ventricular (RV) dysfunction in a third of patients, albeit with preserved functional status.19 Regular follow-up should be routine in patients repaired in adulthood, with residual shunts, elevated PA pressures, or arrhythmias. The risk of arrhythmia, especially atrial fibrillation and flutter, persists following defect closure. However, pre-existing atrial arrhythmias are less prevalent following repair.20
VENTRICULAR SEPTAL DEFECT
Discounting bicuspid aortic valve disease, VSD is the most common CHD, affecting 4.1 per 1,000 live births11 and accounting for 35–40% of CHD worldwide.10 VSDs are usually subdivided into their anatomical location and can be classified as perimembranous, muscular, or subarterial. Most small (usually muscular) VSDs close spontaneously in infancy and early childhood.21 Of the remainder, most are diagnosed and repaired during childhood. As a result, congenital VSDs of haemodynamic significance are rare in adults, with a prevalence of 0.3 per 1,000 population.22
VSDs repaired in childhood without evidence of a residual defect will not need further surgical intervention. Similarly, small VSDs with insignificant effects on the left ventricle (LV) or the pulmonary vasculature usually remain asymptomatic and do not require surgery.23 However, closure is recommended in VSDs with clinical or haemodynamic consequences (Table 1b).
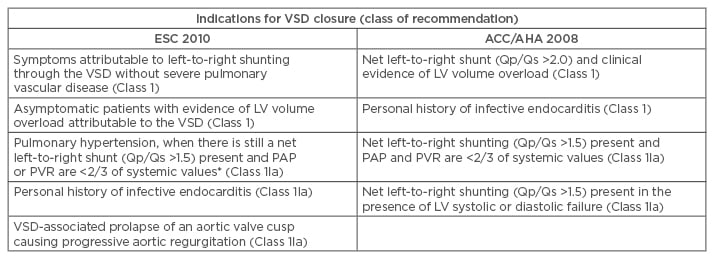
Table 1b: Indications for intervention in ventricular septal defect.
*Measurements at baseline or after vasoreactivity testing or a period of pulmonary hypertension therapy.
LV: left ventricle; PAP: pulmonary artery pressure; PVR: pulmonary vascular resistance; Qp/Qs: pulmonary flow/systemic flow; SVR: systemic vascular resistance; VSD: ventricular septal defect; ACC/AHA: American College of Cardiology/American Heart Association; ESC: European Society of Cardiology.
Adapted from the 2010 ESC guidelines8 and the 2008 ACC/AHA guidelines.9
An Eisenmenger VSD, the classical Eisenmenger’s complex, represents a large defect associated with severe PH, shunt reversal (right-to-left), and cyanosis, and is a contra-indication to closure. In such patients, pharmacotherapy used to treat consequential PH may be beneficial.24-26 A modest amount of randomised controlled trial evidence supports a positive effect on pulmonary haemodynamics, 6-minute walk test distance, and quality of life.27 The effect on survival and the role of combination therapy are unclear.27,28
Surgical techniques for VSD repair have been practiced since the 1950s.29 Surgical closure remains the treatment of choice for most defects.23-35 Surgery is performed through a sternotomy on cardiopulmonary bypass and with transvalvular access to the defect. The closure rate is excellent at ~97–100%.31,34 Technical problems arise where defects cannot be easily accessed, as well as in apical defects and multiple muscular defects. Mortality and major complication rates are low (1–2%), except when elevated PA pressures and RV dysfunction are present.
Catheter-device closure can be used as an alternative to surgery in centrally located muscular VSDs and perimembranous defects.36 It is especially useful in patients with a high-operative-risk, in those with previous attempts at surgical closure, and in restrictive VSD closure. Device closure has a high implantation success rate (96.6%) with relatively few complications. These include a residual shunt (3.1%) and cardiac dysrhythmias (10.6%).37 Complete heart block following percutaneous closure of peri-membranous VSDs occurs in 1% of cases,38,39 and may develop years after repair. Complications are more common in children weighing <5 kg.40
Follow-up is recommended in all patients with a VSD, with shorter interval and tertiary centre follow-up for patients with LV dysfunction, residual shunt, PH, aortic regurgitation, outflow tract obstruction, and evidence of dysrhythmia.8 Adults with small, unrepaired defects require follow-up due to the risk of an increase in shunt size, LV volume overload, and other complications, which may necessitate late repair.41,42 Transthoracic echocardiography is usually sufficient in the surveillance of these patients. Following spontaneous closure of a VSD, or where surgical repair has been successful without a residual defect or associated lesions and with normal PA pressures, routine follow-up in a specialist centre is not required.
Patients with VSDs are at increased risk of endocarditis. Meticulous dental hygeine and regular dental review are strongly encouraged. Antibiotic prophylaxis for prevention of procedure-associated infection is not routinely recommended for uncomplicated VSDs.43 However, the American Heart Association (AHA) recommends antibiotic prophylaxis for dental procedures in certain situations, such as in the 6 months following complete VSD closure and indefinitely in the presence of a residual VSD in relation to patch material.44
PATENT DUCTUS ARTERIOSUS
The ductus arteriosus is a vital structure in the fetus, connecting the proximal left PA and the descending aorta distal to the left subclavian artery. It allows shunting of blood across the non-functioning pulmonary circulation. Its persistence beyond a few weeks after birth is termed PDA. Its prevalence is 2.9 per 10,000 live births,11 representing 10–15% of all adult CHD. PDA usually presents as an isolated lesion in adults, unlike in young children where concurrent complex defects are often present.
The initial pathophysiology is that of left-to-right shunting, the magnitude of which depends on the ductal resistance. This resistance varies with the defect length, narrowest diameter, and shape. The sequelae of LV volume overload and PH depends on the shunt size.
Patients with small ducts without an audible murmur or signs of LV volume overload and with normal PA pressures generally remain asymptomatic. They have a normal life expectancy and do not require repair. The presence of a continuous murmur in a small defect without signs of LV overload should be considered for closure (Class IIa).8
Moderate or large defects may be asymptomatic in childhood, or may present with fatigue and exertional dyspnoea. LV volume overload or PH may predominate and are both indications for closure (Class I). Patients with PH should be considered for closure, even when pulmonary vascular resistance is very high, as long as there is evidence of a net left-to-right shunt or pulmonary vascular reactivity at catheterisation (Class IIa). Adults with large defects may present with Eisenmenger’s syndrome. Here, closure must be avoided (Class III).8,9 The management and outcomes are similar to Eisenmenger VSD.
Where the anatomy permits, transcatheter device closure is the preferred mode of treatment, even when cardiac surgery is indicated for correction of concomitant cardiac lesions. The most frequent method used, transcatheter coil occlusion, was developed in 1992 by Cambier et al.45 Currently, a number of occluder devices exist for different defect morphologies, achieving complete closure rates of 85–100%.46 Serious complications are rare, including device embolisation, flow disturbance from device protrusion, access site thrombosis, and infection.
Surgical repair has been practiced since the first reported successful PDA ligation by Gross and Hubbard47 in 1939. Surgical ligation or division are both effective. In adults, however, aortic and PDA calcification, as well as tissue friability around the aortic isthmus and PA, increase the complication rate, including PDA rupture.48 In older adults, aortic calcification is routinely seen and transcatheter closure is therefore the preferred approach. Consequently, surgery is generally reserved for patients with very large ducts, unfavourable duct morphology for transcatheter closure,49,50 or in accompanying PDA aneurysms (8% of pre-natal cases,51 but rare in adults).
Follow-up is not required beyond 6 months following PDA closure in the absence of a residual shunt, LV dysfunction, or high PA pressures. The presence of a residual shunt, LV dysfunction, or PH necessitates follow-up in a specialist centre at 1–3 yearly intervals.
COARCTATION OF THE AORTA
CoA accounts for 5–8% of all CHD with a prevalence of 4.4 per 10,000 live births.2,11 It is a generalised aortopathy rather than a localised pathology and represents a broad anatomical spectrum of disease; from a mild, discrete narrowing to a long, hypoplastic segment of the aorta. This clinical entity should be distinguished from pseudocoarctation, which is characterised by a kinked, elongated aortic segment without significant obstruction to blood flow (≤20 mmHg at cardiac catheterisation). Bicuspid aortic valve is the most frequent associated lesion, present in 85% of cases.
CoA is classified by its anatomical relationship with the PDA or ligamentum arteriosus and the aortic arch. A pre-ductal CoA is seen in the setting of an infant with a large PDA, where systemic blood flow is dependent on the right-to-left shunting of blood to the descending aorta. This ductal-dependent circulation requires prostaglandins to maintain duct patency until emergency repair is performed. Post-ductal CoA presents depending on the severity of the stenosis, presence of concomitant intra-cardiac lesions, and the presence of collateral vessels. Adults with CoA have usually been repaired in childhood, unless diagnosed in adulthood when investigating early-onset or resistant hypertension. Indeed, the 2008 American College of Cardiology (ACC)/AHA guidelines for adult CHD recommend that all patients with systemic arterial hypertension should be examined for brachial-femoral delay, together with performing supine bilateral arm blood pressures and prone right or left leg popliteal artery blood pressures, looking for differential pressures.9
Timing of repair varies according to severity. In the absence of pre-ductal CoA, early repair of childhood CoA is recommended between 2 and 5 years of age. Indications for repair in adult patients depend on symptoms, examination findings, and imaging criteria.8 Repair is indicated in patients with a non-invasive pressure difference >20 mmHg between upper and lower limbs, with one of the following:
- Upper limb hypertension (>140/90 mmHg)
- Pathological blood pressure response during exercise
- Significant LV hypertrophy (all Class I)
Patients with hypertension and ≥50% aortic narrowing relative to the diameter of the diaphragmatic aorta by computed tomography (CT), cardiovascular magnetic resonance (CMR), or invasive angiography, should be considered for intervention (Class IIa). It is unclear whether an incidental finding of a significant narrowing in the absence of symptoms or hypertension should be repaired (Class IIb).
Surgical repair of CoA was realised in 1944, when Clarence Crafoord52 performed the first successful resection with end-to-end anastomosis in humans, and replicated by Robert Gross in 1945.53 The main types of CoA repair are summarised in Figure 1. Surgery has a low mortality (<1%) in children, but this increases significantly over the age of 30.
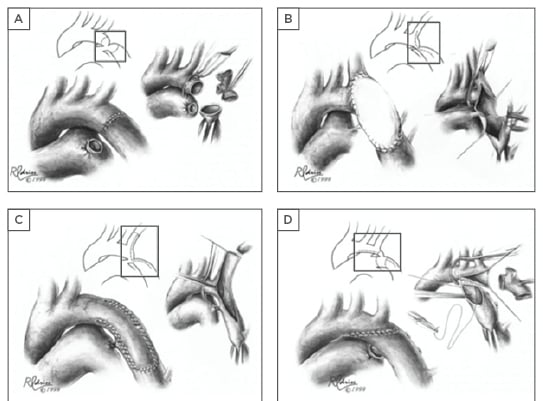
Figure 1: Surgical techniques in coarctation of the aorta repair.
A) Resection and direct end-to-end anastomosis; B) Patch aortoplasty; C) Subclavian flap aortoplasty; D) Resection with extended end-to-end anastomosis. Regardless of the surgical technique applied, late onset complications of re-coarctation and aneurysm formation (particularly following patch angioplasty) should be actively monitored for.
Figures reprinted with permission.54
Percutaneous balloon angioplasty was developed as an alternative to surgical repair in the late 1970s.55 For lasting results, the technique generally involves overstretching of the vessel and a therapeutic tear of the intima with variable extension to the adventitia.56 This predisposes to complications, such as dissection, false aneurysm, and rupture, with a relatively high rate of aneurysm formation (2–20%).57,58 The deployment of aortic stents, both covered and bare-metal, has become the treatment of choice in those with favourable anatomy. CoA stenting overcomes many of the shortcomings of balloon dilatation by scaffolding the target lesion, reducing recoil, and recurrent stenosis. Stents can be deployed without overstretching and tearing, resulting in fewer vessel wall complications. Recent experience with covered stents gives an incidence of aneurysm formation and vessel rupture of <1%.59 In repaired adults with residual or recurring CoA, angioplasty with or without stenting may be effective,60 although surgery remains the modality of choice for recurrent long segment disease, or where there is concurrent hypoplasia of the arch.
Regular follow-up with interval imaging of the aorta is required in all patients. CMR is the preferred imaging modality to reduce lifetime radiation dosing. Measurement of blood pressure and appropriate use of ambulatory blood pressure monitoring is imperative as hypertension is common, even in the absence of significant residual coarctation. Imaging focusses on identifying the post-repair anatomy, associated cardiac lesions, and the development of complications, mainly restenosis and aneurysm formation. Late complications and associated pathology mean that patients with repaired CoA have a shortened life expectancy, with a 20-year survival of 84%.61
PULMONARY STENOSIS
PS accounts for ~8% of all CHD at birth,10 affecting 5.5 per 10,000 live births.11 Obstruction is most common at the valve level as an isolated defect, accounting for 80–90% of cases. RV outflow tract obstruction (RVOTO) also occurs at the subvalvular level (infundibular or sub-infundibular PS; 5%), supravalvular level (1–2%), in a branch PA (discrete, or diffuse ‘hypoplasia’; 5%), or as a combination of the above.62 Taken together, RVOTO and PS may contribute to 20% of all CHD, including as part of tetralogy of Fallot. It is the most common valve lesion that requires therapy in adults, in part due to previously repaired patients presenting with residual lesions, such as pulmonary regurgitation or restenosis.
Morphologically, the valve is dome-shaped in the majority of cases, with a narrow central opening, but mobile valve cusps. In <20% of cases, the valve is dysplastic with poorly mobile cusps. This is characteristic of PS in Noonan’s syndrome, diagnosed during adulthood in 8% of cases.63 Stenotic valves may calcify with advancing age.
Presentation and clinical history varies with the site and severity of the RVOTO. Severe PS, isolated or as part of a syndrome (such as tetralogy of Fallot, truncus arteriosus, or some types of transposition of the great arteries) will present in neonates with heart failure and may be duct-dependent. This necessitates early repair. Most other lesions lead to progressive RVOTO, either through direct valvular calcification or reactive myocardial hypertrophy, which presents with reduced exercise capacity and exertional dyspnoea. Mild valvular PS usually does not progress.64
According to the 2010 European Society of Cardiology (ESC) guidelines,8 indications for repair include RVOTO at any level, regardless of symptoms, where the Doppler peak gradient is >64 mmHg (Class I). Repair should be considered in symptomatic patients, those with RV dysfunction, double-chambered RV (from a small VSD), significant arrhythmias, or right-to-left shunting via an ASD or VSD who do not meet Doppler criteria (Class IIa). Peripheral PS should be assessed for repair if >50% diameter narrowing and RV systolic pressure >50 mmHg with or without lung perfusion abnormalities (Class IIa).
In valvular PS, transcatheter balloon valvotomy is the treatment of choice. The procedure is considered successful when the invasive transvalvular gradient falls to <30 mmHg. The procedure is relatively safe, with the rates of mortality and serious complications cited as 0.24% and 0.35%, respectively.62 Rarely occurring complications include transient bradycardia and hypotension at the time of balloon inflation, right bundle branch block or atrio-ventricular block, PA tears, and balloon malfunction.
Surgical therapy is still necessary for significant residual PS despite repeated balloon valvotomies and for more complex lesions. Bioprosthetic valves are preferred to mechanical valves due to the increased risk of thrombosis in right-sided lesions. More complex lesions may necessitate RVOT reconstruction in the form of patch augmentation and valved conduits. A growing minority of patients requiring intervention are suitable for percutaneous pulmonary valve implantation (PPVI). Where feasible, PPVI is a safe and effective alternative to surgical valve replacement. Follow-up studies ≤7 years have shown a high procedural success rate with a low incidence of post-procedure pulmonary regurgitation and a 5-year re-intervention rate of <25%.65
In patients with previous pulmonary valve intervention, progressive RVOT dysfunction may occur due to several mechanisms, including pulmonary regurgitation and conduit malfunction (stenosis, calcification, and kinking).66 This may lead to several lifetime procedures, with repeated operations being associated with increased morbidity. In this population, PPVI may avoid or postpone open-heart surgery and its associated risks.
Patients with RVOTO require life-long follow-up with interval echocardiographic imaging. Most patients will be seen annually in specialised CHD centres, although those with mild valvular or residual PS require follow-up every 5 years.
CONCLUSIONS
Early diagnosis and, where possible, repair of congenital heart defects has created a population of adults with CHD, which is projected to rise significantly over the coming decades. Repair is rarely ‘curative’, with patients requiring lifelong follow-up for surveillance of late complications.
Furthermore, specialist CHD services are provided by relatively few centres, each covering large geographic areas. Therefore, adults with repaired and native congenital heart defects may present to local hospitals staffed by non-specialists, either with a chief cardiovascular complaint or with a cardiac co-morbidity. Consequently, a basic understanding of the anatomy, physiology, and the potential therapeutic interventions for these conditions is necessary to be able to make an effective assessment, provide urgent treatment and communicate with the tertiary centre as needed.
In a ‘hub-and-spoke’ approach, local units may be supported in looking after patients with simple defects. Care of patients with moderate-to-complex lesions requires closer links and referral to specialist centres which requires effective national frameworks combined with widespread education programmes tackling the most frequently encountered congenital heart defects.