
Abstract
Personalised medicine, with the aid of companion diagnostics, is a burgeoning field. The potential benefits of personalised medicine with regard to improved patient outcomes and reducing healthcare burden are recognised, but there remains obstacles that may limit growth in this area. Limitations include the current regulatory framework in many areas, in which the pharmaceutical is identified as a medicine, whilst the companion diagnostics are identified as a medical device; thus the two components may be governed and assessed by differing bodies and processes. This in turn results in disparity in approval times, patent and intellectual property claims, and reimbursement. Regulatory agencies are working together with industry and academia towards bridging these gaps, with significant inroads seen across the globe.
INTRODUCTION
In recent times, the development of personalised medicine has been amongst the most significant outcomes of the acceleration in genomic science.1 This development has deepened our understanding and ability to predict both the process of disease evolution and the mechanisms of action through which potential therapies can interrupt or arrest this evolution.2 The concept of personalised medicine, or precision medicine, is to tailor a treatment regime for the individual to achieve optimal results, based on a detailed understanding of the molecular and genetic basis of diseases. This has a number of advantages, including: avoiding unnecessary treatment that would be of little to no benefit and a closer understanding of the risk-to-benefit ratio for a patient, thus ensuring that the risk of adverse drug reactions in that particular patient are likely to be outweighed by the benefits.
Two broad categories of diagnostics have emerged with the advent of personalised medicine: complementary and companion diagnostics (CDxs). Complementary diagnostics are generally identified as assays that inform on the potential benefit of a therapeutic for an individual, but do not require a regulatory link to a specific pharmaceutical at the time of development. In contrast, CDxs are directly linked to a specific pharmaceutical during and post-regulatory approval.3 CDxs identify those individuals with expression of specific genetic biomarkers that identify a disease or the likelihood of response to a therapy, as well as tools to optimise and monitor therapeutic doses. The potential benefits of this type of treatment regime are numerous, including the optimisation of treatment, decreasing healthcare costs by treating only those patients that will benefit from the therapy, which is associated with a reduction in waiting times for treatments; reduction to treatment delay with a second-tier treatment that may be of greater benefit to a patient than the primary treatment; and focussed clinical development of the therapeutic, enhancing the risk-to-benefit profile, which is advantageous for patients, industry, and for regulatory approvals and compliance.4 However, there are also a number of stumbling blocks and disappointments, as is the case with most new paradigms, including patient and societal expectations, which may be unrealistic in terms of both improved efficacy of the treatment and increased ability to treat a greater number of disorders, costs, and resources associated with undertaking CDxs,5 patent associated issues resulting in the reluctance of companies to invest in research and development of the CDx,6,7 differential reimbursement/payment schemes for the pharmaceutical and the CDx,8 and traversing the regulatory landscape whilst it is under development to accommodate the interplay between pharmaceutical and CDx.
The development of CDxs is ideally concomitant with a pharmaceutical, thus enabling appropriate evidence collection to demonstrate and evaluate the risk-benefit profile of particular patient cohorts. Previous research has identified the need for the development of new models of research and development to address the area of CDxs.9 However, it is also essential to consider the emerging challenges for both industry and regulators in the co-ordinated approval and post-market monitoring of these medicines and medical devices.
CURRENT STATUS OF APPROVED COMPANION DIAGNOSTICS
The CDx is, in essence, similar to any other in vitro diagnostic (IVD) device in regard to the purpose of detecting the presence of an analyte or genetic sequence to inform on the potential susceptibility to a disease, potential efficacy of a therapeutic, or in monitoring the effectiveness of a treatment regime. Similarly, the evidence supporting the intended purpose of the device is similar to that of other medical devices. The differentiator for a CDx from other IVD assays is the association specified within the approved labelling of a particular pharmaceutical. Examples include the 26 CDxs currently cleared or approved by the US Food and Drug Administration (FDA), in which the intended use specifies the associated treatment (Table 1).
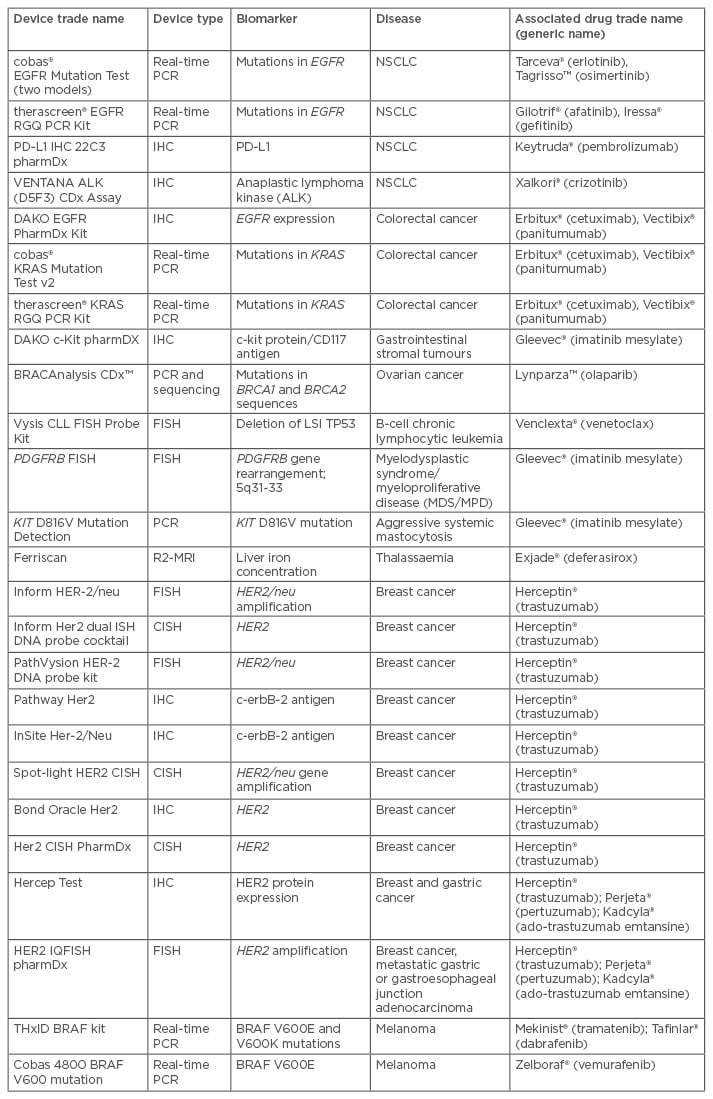
Table 1: Companion diagnostic devices cleared or approved by the US Food and Drug Administration (FDA).14
EGFR: epidermal growth factor receptor; ALK: anaplastic lymphoma kinase; NSCLC: non-small cell lung cancer; PCR: polymerase chain reaction; IHC: immunohistochemistry; MRI: magnetic resonance imaging; CISH: chromogenic in situ hybridisation; FISH: fluouresence in situ hybridisation.
Interestingly, some of these pharmaceuticals have benefited with the advent of CDxs. Iressa® (gefitinib) and Herceptin® (trastuzumab) are examples of pharmaceuticals that were not considered cost-effective or sufficiently efficacious in cancer treatment prior to being coupled with a CDx. In the case of gefitinib, in 2005, following failure to demonstrate significant benefit in Phase III clinical trials, use was restricted in the USA market and the marketing authorisation application was withdrawn in Europe. However, an additional Phase III trial identified that the patient cohort with an epidermal growth factor receptor gene (EGFR) mutation had a greater favourable outcome when treated with gefitinib, resulting in the European Medicines Agency (EMA) approving treatment of gefitinib, with the CDxs for EGFR-tyrosine mutation, in non-small cell lung cancer (NSCLC).10
Similarly, when trastuzumab was assessed for the treatment of advanced gastric cancer by the National Institute for Health and Clinical Evidence (NICE), an independent body which provides guidance to the UK National Health Service (NHS) on the clinical and cost-effectiveness of health technologies, it was initially not recommended on the basis of the latter. However, following a resubmission of health economic data with the patient subset of HER2 overexpression being defined, the outcome was positive.11 This example not only highlights the importance of defining the optimal patient population for the therapeutic, but also the benefit of health economic reviews where the cost and value of the CDxs and pharmaceutical are jointly assessed to clearly demonstrate the cost-effectiveness.
Additional benefits of understanding the disease process and drug mechanistics, and thus in turn the effect on different patient subpopulations, are reflected in terms of decreased development times and improved approval process for new pharmaceuticals. For example, the development and approval of Zykadia® (ceritinib) in the treatment of NSCLC was based on small open-labelled non-randomised Phase I/II studies in patients with anaplastic lymphoma kinase rearrangement who had developed resistance to Xalkori® (crizotinib). The patient cohort with the specific genetic marker and CDxs had already been indentified with the development and approval of crizotinib.12,13
CHALLENGES FOR REGULATORY APPROVAL
Regulatory agencies, in many first-world jurisdictions, identify a CDx as a medical device and a pharmaceutical therapeutic as a medicine. This in turn requires separate applications through two separate regulatory frameworks. The differing requirements of these two regulatory systems may result in a delay in the approval of one component. The outcome may be a pharmaceutical that cannot be prescribed, despite the benefit identified, as the CDx lacks approval, resulting in a delay in patient treatment. Conversely, a diagnostic tool may be approved prior to the medicine, resulting in a CDx that has no purpose, as the associated pharmaceutical has not been approved. Whilst the latter may not appear to be of significant consequence clinically, it does have a substantial financial impact for the manufacturer of the CDx who has invested extensively in the development and validation of the tool. In addition, the early release of a CDx may result in ‘fast-follower’ devices being brought to market without the same investment, in part due to the inability of developers to patent these types of devices.4,6 This in turn has an impact on the willingness of industry to invest in this area, which may lead to stagnation of innovation and an impediment to the development of personalised medicine.
Additional challenges that may be faced when seeking regulatory approval of personalised medicines include the difficulty in undertaking clinical trials of sufficient size when the patient cohort is relatively small. This issue is further compounded when the therapy is highly personalised, such as RNA-based pharmacotherapies.15 Consideration must also be given to the regulatory requirements relating to the inclusion of biomarker-negative patients in clinical trials, with exclusion and inclusion criteria differing across regulatory agencies. Moreover, there are challenges and uncertainties with the introduction of alternate biomarkers, and therefore a new CDx, which may also alter the patient apparent designation as either biomarker-negative or positive, dependent upon the marker.16
Other challenges arise from the differences in international dossier requirements for the submission of a medicine and medical device and involvement of different bodies within countries reviewing each dossier. For example, in Europe a medical device dossier may be reviewed by a notified body, whereas a pharmaceutical dossier may be reviewed by the EMA. From a regulatory perspective, attention must also be given to potential loop-holes in regulatory frameworks. Previous research has documented issues with the utilisation of laboratory-based diagnostic tools that do not undergo equivalent scrutiny as a CDx.17,18 This raises concern for both the consistency of approval and the ability to ensure post-market safety of both the medical device and its accompanying pharmaceutical agent.
Despite all the challenges, there are examples where the co-ordinated approval of both the CDx and the pharmaceutical has occurred concurrently, and furthermore, approval has been expedited. For example, under the US FDA’s priority review programme, the approval of Zelboraf®, in conjunction with the cobas 4800 BRAF V600 mutation CDx, was expedited for the treatment of metastatic or unresectable melanoma.19 Both manufacturers and regulatory agencies should reflect upon exemplars such as this to better inform policy and practice.
CHALLENGES FOR POST-MARKET MONITORING AND REGULATION
Once approved, manufacturers, sponsors, and regulatory agencies must then employ appropriate post-market monitoring of both the pharmaceutical and medical device. This also raises challenges, for example: integration of pharmacovigilance data into the quality management system of a medical device, integration of device vigilance data into pharmacovigilance tools such as a periodic safety update report, and combined reporting of medicine and medical device adverse events.
Furthermore, the ability to adequately implement regulatory actions, such as recalls and safety alerts for a companion medical device must be considered. This can have a significant impact on the ability to administer and monitor the accompanying pharmaceutical, including the potential impact of delayed or interrupted therapy cycles. Appropriate systems to communicate and manage the risk of post-market problems with either the medicine or medical device component must be considered and documented.
NAVIGATING THE CHALLENGE: EMERGING PATHWAYS
To overcome issues with disparity between the pre-existing regulatory processes of medical devices and medicines, regulatory agencies have been consulting with academia, industry, and other international regulatory agencies, to explore a streamlined approach to regulating medicines with their CDx.16 Despite many regulatory agencies still having separate approval and post-market vigilance areas for medicines and medical devices, with no evidence of an integrated approach being implemented, guidance documentation, draft regulations, and legislative proposals are now becoming available that reference personalised medicine (Table 2). In addition, there are initiatives being implemented with the aim of bridging the gaps between the separate regulatory areas, including proposals such as the central Medical Device Coordination Group (MDCG) proposed in Europe. The working group is proposed to consist of experts in medical and IVD devices, to assess high-risk and CDx devices, working in conjunction with designated reference laboratories, notified bodies, and the EMA.20 Whilst many regulatory agencies recognise the benefit of personalised medicine, as well as the challenges, countries such as Japan and the USA appear to be leading the way in terms of pre-market approval processes.
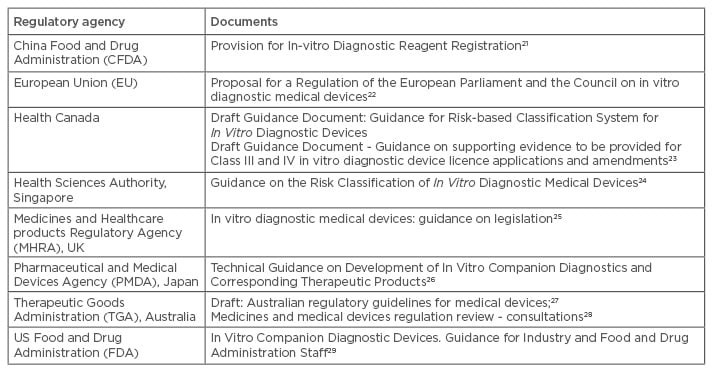
Table 2: Exemplar regulatory guidance documents.
The development of guidance documents or amendments to legislation to keep up with health innovations, such as companion diagnostics, is varied across the globe.
CONCLUSION
The benefit of personalised medicine is clearly evident, not only to the patients, but also to the healthcare system and pharmaceutical companies both pre and post-regulatory approval. The need for CDxs, which are appropriately sensitive, specific, and accurate, is similarly evident. It is in the regulatory space, and some would argue the reimbursement and patent areas also, where there is still work to be done to provide legislation and guidelines that meld the approval systems for the medicine and CDx, which will ultimately benefit not only the manufacturer, but also patients and regulatory bodies. However, this does not necessarily equate to faster process times of applications, nor a relaxation in the scrutiny of review that is undertaken, but rather a way to streamline applications and bring together regulatory oversight of the medicine and medical device, in both the pre and post-market arena.






