
Abstract
The scientific community has made great strides in responding to the huge public health problems of obesity and diabetes with the discovery of the incretin system and the development of glucagon-like peptide 1 analogues. These have shown clinical efficacy in randomised controlled trials and observational data from real-world evidence; however, a ‘treatment gap’ remains between the therapeutic success of these molecules and the outcomes achieved with bariatric surgery. To help address this, dual incretins are being developed. These combine glucagon-like peptide 1 action with that of either glucose-dependent insulinotropic peptide or glucagon. This narrative review charts the development of incretin therapy, and the dual agonists for treatment of Type 2 diabetes and obesity.
Key Points
1. Agonists of the incretin glucagon-like peptide-1 (GLP-1) have established efficacy for glycaemic reduction, weight loss, and cardiovascular risk modification; however, some patients may still not achieve metabolic targets and efficacy remains considerably less than that seen with bariatric surgery.2. In recent years, dual agonists of the incretin system have been tested in pre-clinical and early-phase clinical trials, with the glucose-dependent insulinotropic polypeptide and GLP-1 co-agonist, tirzepatide, recently approved for clinical use. This provides near double the weight loss to GLP-1 agonists and significant glucose lowering, as well as promising efficacy in non-alcoholic fatty liver disease. Future prospects include triple agonists of glucagon, glucose-dependent insulinotropic polypeptide, and GLP-1.
3. This article highlights the evolving understanding of the incretin system over the years, leading to the development of multi-agonists, which hold the potential for marked metabolic benefits, mimicking that seen with bariatric surgery.
INTRODUCTION
The identification of the incretin system as consisting of hormones located in the gastrointestinal mucosa, that have insulinotropic action; and its subsequent exploitation in the form of glucagon-like peptide 1 (GLP-1) receptor agonists (RA) has been a therapeutic success story for Type 2 diabetes (T2D) and obesity management. Nonetheless, therapeutic failure, partly from inability to maximise the dose or persist with the medication because of nausea and vomiting, limits the potential of this class of drugs. The recognition of the contribution of other hormones in the incretin system, with pleiotropic actions, has led to the development of dual agonists, which can maximise metabolic benefits with a lower potential for side-effects. This may help to close the ‘treatment gap’ between the effects of existing drugs and that of bariatric surgery on weight and metabolic parameters.
This narrative review covers the identification and development of incretin therapy for diabetes and obesity, culminating in the recent clinical trials of dual agonist therapy and the future prospect of tri-agonists.
SEARCH METHODOLOGY
The authors searched the English-language literature to identify all relevant studies, regardless of publication status or year of publication. They searched the PubMed and Google scholar databases, combining the terms dual agonist’ OR ‘triple agonist’ OR ‘GIP/GLP-1’ OR ‘[glucagon-like peptide 1]’ PLUS ‘[glucose dependent inhibitory peptide]’ OR ‘glucagon/GLP-1’ OR ‘dual incretins’. Studies could include pre-clinical or early or late phase human trials. Additional studies were added by cross referencing the retrieved studies. The last search was performed in July 2022.
DISCOVERY OF INCRETINS
The superiority of enteral, compared to parenteral glucose administration in stimulating insulin secretion in humans, was first described in 1964 by McIntyre et al.1 This paper reignited long-dormant interest in the concept of incretins: putative gastrointestinal factors released during meal absorption that might stimulate insulin secretion in a glucose-dependent fashion. Two hormones released by enteroendocrine cells in the gut mucosa were subsequently discovered to be largely responsible for the incretin phenomenon. Glucose-dependent insulinotropic polypeptide (GIP, also known as gastric inhibitory polypeptide) was the first to have its incretin effect demonstrated, by the Canadian team of Dupre et al.2 Identification of the other hormone, GLP-1, was a more complex task because of the difficulty of differentiating it immunologically from other bioactive products and inactive degradation fragments of its precursor, proglucagon. The ability of intravenously infused GLP-1 to stimulate insulin secretion in humans in a glucose-dependent fashion was eventually described by Kreymann et al.3
GLUCAGON-LIKE PEPTIDE-1
Despite being identified later than GIP, GLP-1 quickly became the focus of intensive research, such that fewer than 20 years were to elapse between the description of its incretin effect and the approval of the first highly successful GLP-1 RA drug class for treatment of T2D.4 GLP-1 is released after food intake3 into the enterohepatic portal system by L cells located predominantly in the mucosa of the ileum and large bowel.5 The bioactive forms, GLP-1(7–37) and most abundantly, GLP-1(7–36)NH2, are inactivated by dipeptidyl peptidase-4 (DPP-4).6 Intravenously infused GLP-1 slows gastric emptying,7 reduces appetite and food intake,8 and suppresses glucagon (GCG) secretion at normal and high blood glucose concentrations, but not during hypoglycaemia.9 Further evidence for the physiological role of GLP-1 in regulating GCG secretion accrued from studies showing that exendin (9–39), an antagonist of the GLP-1 receptor, impairs oral glucose tolerance10 by disinhibiting GCG secretion when blood glucose concentration is normal or elevated.11 As a result, GLP-1 RAs reduce hyperglycaemia with little risk of hypoglycaemic episodes.
Development of Drugs Acting on the Glucagon-Like Peptide-1 Receptor
Exendin-4, a peptide component of the venom of the Gila monster (Heloderma suspectum), has approximately 50% structural homology to GLP-1(7–36)NH2 and is a DPP4-resistant agonist at the GLP-1 receptor (GLP-1R).12 As this confers a much longer half-life than that of native sequence GLP-1 (hours rather than minutes), the development of GLP-1 RAs has proceeded down two paths: as analogues of either human GLP-1, or of exendin-4.
Human GLP-1-analogues that have come to market include liraglutide, dulaglutide, semaglutide, and albiglutide (the last has been withdrawn). Marketed drugs based on exendin-4 are exenatide and lixisenatide (Figure 1).12,13 Modifications to prolong the half-life of GLP-1 RAs include sequence substitutions conferring resistance to DPP-4, along with acylation (liraglutide and semaglutide), covalent bonding to modified Ig fragment crystallisable domain (dulaglutide), or covalent bonding to albumin. These various approaches delay clearance by the kidney.
Notwithstanding their resistance to degradation by DPP-4, exendin-4-based GLP-1 RAs require daily (lixisenatide) or twice daily (exenatide) dosing without further modification. A weekly version of exenatide (Bydureon [AstraZeneca, Cambridge, UK]) has therefore been developed, in which co-formulation with poly(D,L-lactide-co-glycolide), a material similar to that used in soluble sutures, slows release of drug from the injection site depot.12,13
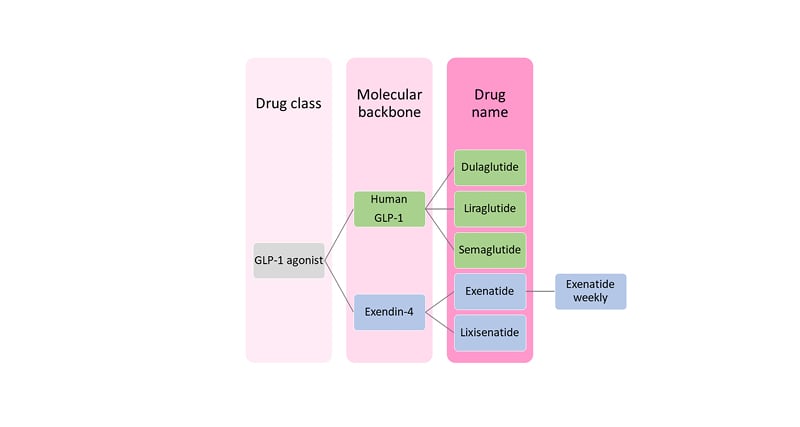
Figure 1: Classification of glucagon-like peptide 1 receptor agonists.
GLP-1: glucagon-like peptide 1.
Regulatory approval for the first GLP-1 RA drug for the treatment of T2D was issued in 2005 and since then GLP-1 RAs have proved highly clinically effective.12,13 Although the original approval was largely based on the potent glucose-dependent insulinotropic properties of GLP-1R activation on pancreatic β-cells, it has become clear that the GLP-1R is expressed on a variety of other tissues, which produce a range of biological effects across diverse organ systems (Figure 2).14
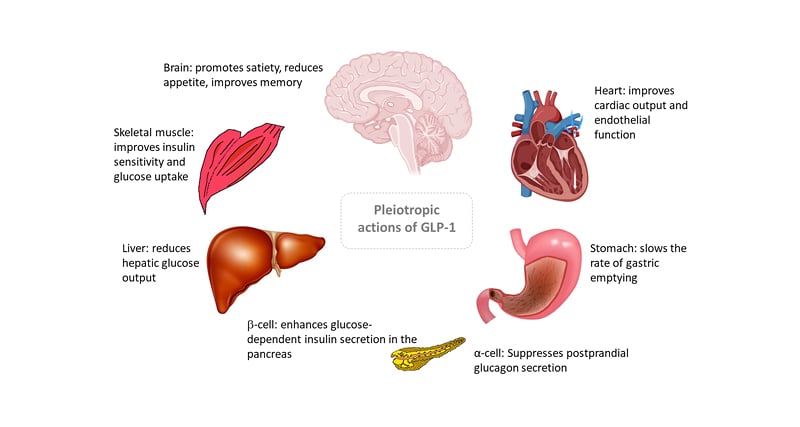
Figure 2: Pleiotropic actions of glucagon-like peptide 1.
GLP-1: glucagon-like peptide 1.
Early research on the therapeutic potential of GLP-1 was directed towards glucose regulation in diabetes but was extended, through concurrent observations of body weight lowering to obesity management. Weight reduction may result from activation of vagal and cerebral GLP-1Rs, leading to appetite suppression. Average weight loss achieved with GLP-1 RA at doses suitable for treating T2D is around 5 kg.12,15 GLP-1 RAs have been licenced (at higher doses than those used for diabetes) for the treatment of obesity. The European Medicine Agency (EMA) approved daily liraglutide (3.0 mg) and weekly semaglutide (1.7 or 2.4 mg) for this indication in 2021.
Studies Showing the Efficacy of Glucagon-Like Peptide-1 Receptor Agonists
In addition to lowering HbA1c, GLP-1 RAs improve traditional cardiovascular disease risk factors such as obesity and hypertension, whilst also exerting anti-inflammatory and anti-atherosclerotic effects, and having positive direct modulatory effects on endothelial function.12 Several large, randomised trials have shown that GLP-1 RAs reduce major adverse cardiovascular events (MACE) compared with placebo. These include LEADER (liraglutide), REWIND (dulaglutide), and SUSTAIN-6 (semaglutide);12,16 however, no statistical differences in MACE outcomes were found in ELIXA (lixisenatide) or EXSCEL (exenatide). Meta-analysis of renal outcomes shows GLP-1 RAs reduce renal events by 15%, primarily through prevention of albuminuria, and mortality by 11%, with no difference between agents;12,16 however, there is a lack of data comparing these GLP-1 RA outcomes versus other glucose lowering therapies.
COMPARATIVE EFFECTS OF GLUCAGON-LIKE PEPTIDE-1 AND GLUCOSE-DEPENDENT INSULINOTROPIC POLYPEPTIDE
Both GLP-1 and GIP have profound effects on energy metabolism and body weight (Table 1). Whilst both have significant incretin effect on insulin secretion, their effects on GCG release differ. GLP-1, but not GIP, slows gastric emptying, which further reduces post-meal glycaemic increments. As described above, GLP-1 inhibits appetite and food intake, resulting in weight loss upon chronic administration; however, GIP is generally thought to have no effects on food intake.17 GIP, but not GLP-1, increases triglyceride storage in white adipose tissue via stimulation of insulin secretion, and through vasodilation of adipose vascular tissue, as well as inhibiting bone resorption. GIP is, therefore, an anabolic hormone that inhibits lipolysis, and stimulates lipogenesis and bone formation. Increased GIP levels are observed in obesity, as fat is a potent stimulus of GIP secretion.17
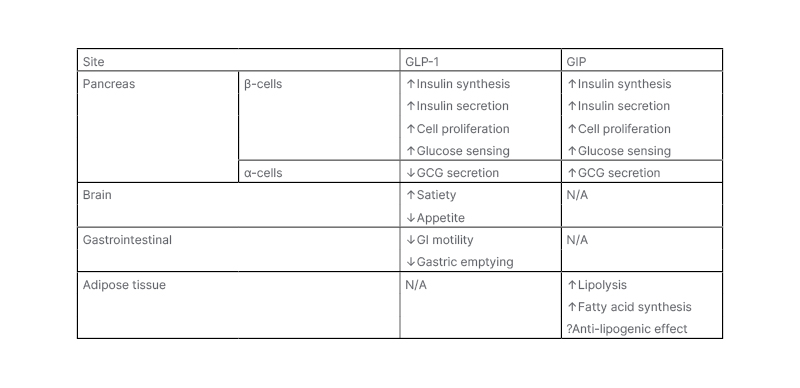
Table 1: Metabolic effects of glucose-dependent insulinotropic peptide and glucacon-like peptide 1.
GCG: glucagon; GI: gastrointestinal; GIP: glucose-dependent insulinotropic peptide; GLP-1: glucagon-like peptide 1.
GLUCOSE-DEPENDENT INSULINOTROPIC POLYPEPTIDE
There was little enthusiasm for therapeutic use of GIP in the years after its discovery because GIP infusions had little effect on insulin secretion and appetite regulation in T2D.17 GIP stimulates GCG secretion and potentiates insulin secretion during hyperglycaemia (Figure 3A);17 however, the insulinotropic effect of GIP is severely curtailed in T2D.18,19 Yet, the glucagonotropic effect of GIP prevails, even during hyperglycaemia (Figure 3B).20 Combined infusions of GIP and GLP-1 have additive insulinotropic effects in the non-diabetic state,21 and resistance to GIP-induced insulin secretion in T2D can be reduced by a short period (weeks) of intensive glucose-lowering treatment (Figure 3C).17,22, This provides a rationale for using GLP-1 together with GIP agonism, as GLP-1 may lower glucose sufficiently to unmask the incretin effect of GIP, while GLP-1 action may overcome the potentiating effect of GIP on GCG secretion. Initial disinterest in the use of GIP has abated and the most prominent, recent approaches to dual incretins are unimolecular combinations of GIP with GLP-1R agonism.
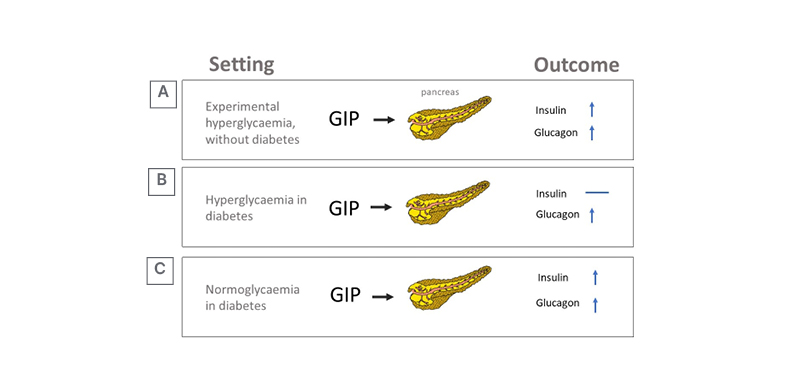
Figure 3: Effect of hyperglycaemia and diabetes on glucose-dependent insulinotropic peptide’s insulinotropic action.
A) Experimental (acute) hyperglycaemia. B) Hyperglycaemia in established diabetes. C) Sustained normoglycaemia in diabetes.
GIP: glucose-dependent insulinotropic peptide.
RATIONALE FOR DUAL OR TRIPLE AGONISTS
As described above, GLP-1 RA have proven efficacy and their use has increased steadily since their launch in 2005.12-14 They are effective for glycaemic control and lead to weight loss that far exceeds that of the intestinal lipase inhibitor, orlistat;23 however, there remains a significant treatment gap in weight management between that attained with GLP-1 RA and bariatric surgery (Figure 4). The human body’s energy balance is governed through much more than a single hormone and it would be anticipated that combinatorial approaches will help close the treatment gap for obesity. Furthermore, the tolerability of GLP-1 RA is often dose-limiting. The most widely reported clinical side-effect of GLP-1 therapy is nausea, which is seen in 13–36% (lower incidence in once weekly preparations).13 Drug surveys have shown that most patients remain at the starting dose.24 There is a need for drugs with equal or better efficacy and a side effect profile that allows dose maximisation. By combining hormones in this way, the dose of individual hormones can be reduced, widening the therapeutic window, and avoiding toxicity.25
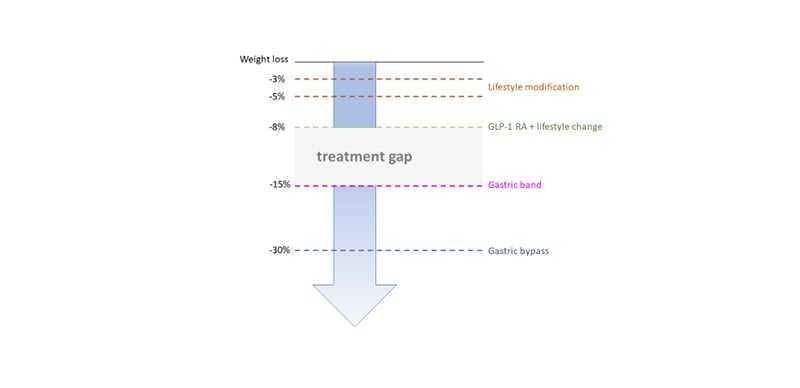
Figure 4. Treatment gap in obesity therapy.
GLP-1 RA: glucagon-like peptide 1 receptor agonists.
Glucagon-Like Peptide-1 and Glucose-Dependent Insulinotropic Polypeptide Dual Agonists
A molecule that attracted early interest was NNC0090-2746 (also known as MAR709, RO6811135, and RG7697). This is a balanced GIP and GLP-1 RA, the development of which has been pursued, unsuccessfully to date, by several pharmaceutical companies.26,27 A Phase II trial in T2D showed a 1% reduction in HbA1c (Diabetes Control and Complications Trial units) and approximately 1.7% body weight reduction over 12 weeks, in comparison to placebo, but a further comparison to an open-label liraglutide active treatment control group was less impressive.26
The drug that has advanced furthest through development, in fact all the way to market, is Eli Lilly and Company’s (Indianapolis, Indiana, USA) tirzepatide (LY3298176), a unimolecular dual GIP/GLP-1 RA. It has comparable GIP receptor binding affinity to native GIP but five-times lower GLP-1R affinity than native GLP-1 and is thus an imbalanced co-agonist.28 This imbalance may allow maximisation of effect on the GIP pathway while minimising GLP-1-related tolerability issues; however, a further possibility has recently come to light. Experiments in islet cell models have shown that intracellular signalling after GLP-1R agonist binding occurs via two pathways, one of which (cyclic adenosine monophosphate generation) enhances insulin release, while the other (β-arrestin recruitment) causes GLP-1R receptor internalisation, and hence reduces cellular sensitivity to ligand.29 Intriguingly, the intracellular consequences of tirzepatide binding at the GLP-1R seem to be peculiarly biased towards cyclic adenosine monophosphate generation, at the expense of β-arrestin recruitment. However, tirzepatide also has low efficacy to induce GLP-1R internalisation, perhaps by its limited ability to recruit β-arrestin. Therefore, the GLP-1R-mediated insulinotropic effect of tirzepatide might be enhanced not just by GIP receptor activity but also by bias in its interaction with the GLP-1R.29
Tirzepatide consists of 39 amino acids, with a C20 unsaturated di-acid acyl chain. The side chain binds to albumin, thereby prolonging the half-life, allowing weekly dosing. Phase II findings published in 2018 were promising, with tirzepatide showing dose-dependent effects on glucose levels and bodyweight.30 It outperformed dulaglutide 1.5 mg/day at the highest doses, albeit in a small group of study participants.
The Phase III trials have been testing tirzepatide as monotherapy, as an add-on to other treatments, and against established glucose-lowering drugs in people with T2D, as well as a weight-loss agent in people with diabetes and obesity. The clinical trial data available have yielded impressive results on glucose control and weight loss (Table 2).31-41 It may be that improvements in islet function and insulin sensitivity with tirzepatide (compared to GLP-1) account for the greater benefits in glucose regulation.42 Cardiovascular effects are being examined in the SUMMIT trial.38 This will test whether people with obesity plus heart failure with preserved ejection fraction, randomised to tirzepatide or placebo for 52 weeks, improve a composite endpoint of mortality, heart failure events, exercise capacity, and heart failure symptoms.43 As a result of the evidence from Phase III trials, the U.S. Food and Drug Administration (FDA)-approved tirzepatide (Mounjaro™ [Eli Lilly and Company]) for the treatment of adults with T2D in May 2022.
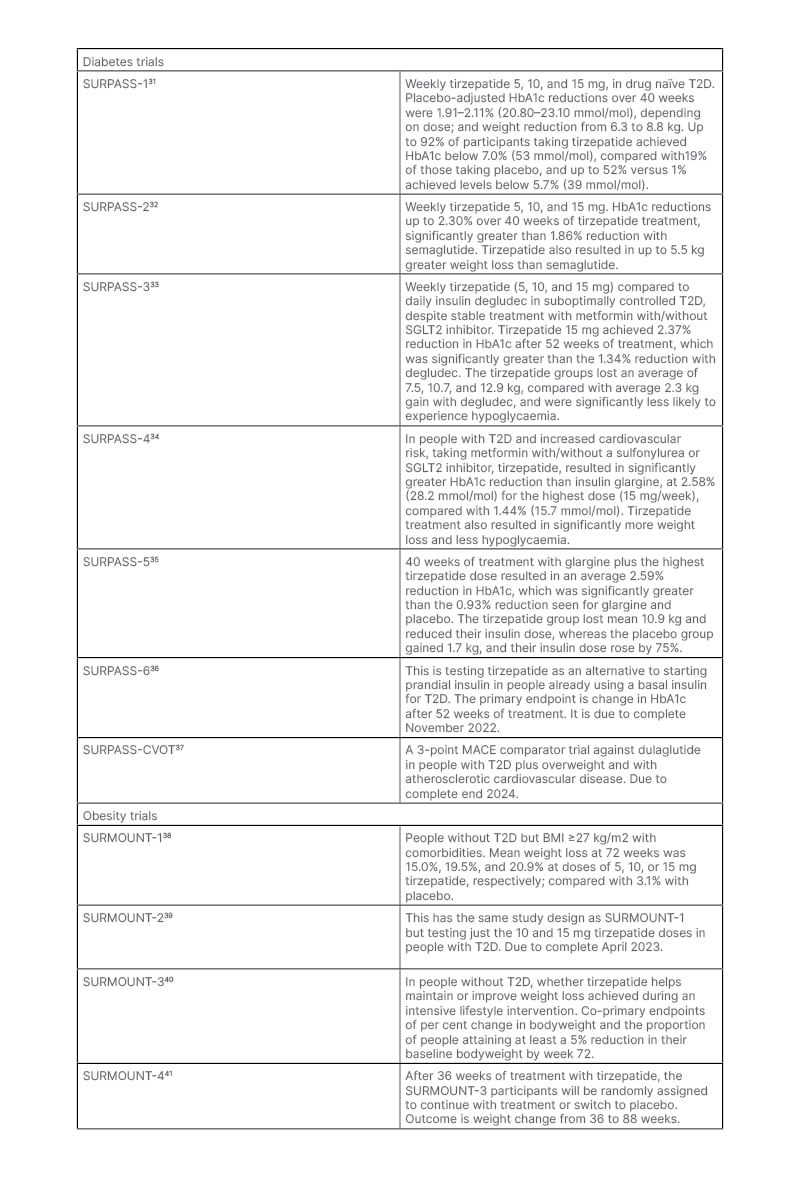
Table 2: Clinical trials of tirzepatide.
MACE: major adverse cardiovascular events; SGLT2: sodium-glucose co-transporter-2; T2D: Type 2 diabetes.
Glucagon-Like Peptide-1 and Glucagon Dual Agonists
The well-known action of GCG is to increase blood glucose (hence ‘glucagon’ being a portmanteau of ‘glucose agonist’). It does so through stimulation of gluconeogenesis and glycogenolysis in the liver. GCG has additional effects including the modulation of food intake and satiety, lipolysis, fatty acid oxidation, ketogenesis, and increased energy expenditure.44 These latter metabolic effects make GCG attractive as an anti-obesity agent, but the applicability, especially in T2D, is complicated by the inherent risk of inducing hyperglycaemia.
Further characterisation of the GCG-like family of peptides led to the discovery of an endogenous GLP-1/glucagon co-agonist ‘oxyntomodulin’, named for its potent effect on stimulating oxyntic (parietal) cells of the stomach to produce gastric acid.45 Oxyntomodulin is co-produced by intestinal L cells in the jejunum, along with GLP-1, and in the colon, in response to nutrient ingestion. Whilst there appears to be no specific oxyntomodulin receptor, endogenous oxyntomodulin causes appetite suppression, increased energy expenditure, and weight loss in people with obesity via activation of GLP-1 and GCG receptors.46 Native oxyntomodulin also significantly augments glucose-dependent insulin secretion acutely in subjects with obesity, with and without diabetes.46 Oxyntomodulin and GLP-1 are increased following bariatric surgery, and contribute to the weight loss and improved glucose control seen after bariatric surgery. GLP-1 and GCG are products of the proglucagon gene, arising from tissue-specific post-translational processing, and share a large degree of sequence homology.47 Co-agonism can, therefore, be obtained by modifying a relatively small number of amino acids from a GCG or GLP-1 backbone. In a seminal study by Day et al.,48 several co-agonists with different GLP-1-to-GCG receptor ratios were investigated in murine models and the optimal balance was suggested to be a co-agonist with equal potency on both receptors. Based on the promising preclinical results, pharmaceutical companies have taken GLP-1/GCG co-agonists into clinical trials.
Mazdutide (also known as IBI362 or LY3305677) is an oxyntomodulin analogue being developed by Eli Lilly and Company. A Phase IB study showed that it was well-tolerated and weight loss up to 6.4% was achieved over 12 weeks.49 The glucose-lowering potential of cotadutide (a GLP-1/GCG RA) was seen in a Phase IIa study, in which the post-prandial area under curve was significantly reduced, and 3.4% weight loss was achieved after 49 days, in adults who are overweight with T2D.50 Efinopegdutide (also known as HM12525A or JNJ-64565111) is a once-weekly GLP-1/GCG co-agonist. In a dosing ranging study, 472 individuals who are overweight without diabetes were randomised and 72% completed 26 weeks of treatment. Weight loss of -6.8%, -8.1%, and -10.0% were seen at 5.0 mg, 7.4 mg, and 10.0 mg doses, compared to -5.8% with liraglutide 3.0 mg;51 however, nausea and vomiting were higher with efinopegdutide. A niche for this drug may prove to be the treatment of non-alcoholic steatohepatitis, as the effects of GCG agonist on hepatic fat oxidation would be desirable in this group.
TRIPLE AGONISM
Having determined the therapeutic potential of the dual co-agonists (with either GCG or GIP complementing GLP-1), Finan et al.52 showed proof of concept that simultaneous agonism at all three receptors, through a single molecule, could be achieved and produce superior therapeutic outcomes.
In a series of experiments in monkeys with obesity and diabetes, the tri-agonist SAR441255 showed superior metabolic outcomes to that achieved with a comparator dual GLP-1/GCG RA.53 Subsequently, Phase I experiments reported that the drug was well tolerated; and the simultaneous receptor engagement, that had been seen in monkeys, of GIP, glucagon, and GLP-1 was confirmed in humans.53
Another GIP/GLP-1/GCG triple RA, LY3437943, has also undergone Phase I trials. After 3 months of treatment, a placebo-adjusted decrease of HbA1c up to 1.56% was seen, with placebo-adjusted body weight reduction of -8.96 kg.54 Data from two larger, Phase II trials are due in the coming months.
Another molecule, HM15211 is a triple full agonist chemically conjugated with a constant region (fragment crystallisable) of human Ig, allowing weekly dosing. As with GLP-1/GCG dual agonists, these triple agonists may have differential efficacy in non-alcoholic steatohepatitis, through reduction in energy intake, β-oxidation, and de novo lipogenesis. In preclinical models, liver fat and liver enzymes were both reduced by LY3437943 and by HM15211.47
CONCLUSION
There is a promising future for the treatment of T2D, obesity, and its metabolic complications. Harnessing the full potential of the incretin system with dual, or possibly triple agonists, appears to produce incremental metabolic gains. This is possible without furthering, often lessening, the side effect profile of GLP-1 RA.
The GLP-1 RA are still under prescribed in developed and well-funded health care systems and are not even available in the most modern formulations in some parts of the world. The authors hope that this review will contribute to a better understanding of the role and relevance of GLP-1 RAs in the treatment of T2D.






