
Abstract
Alpha-1 antitrypsin (AAT), encoded by the SERPINA1 gene, is a protein mainly produced and secreted by hepatocytes. Some specific mutations affecting SERPINA1 may cause accumulation of misfolded AAT in the endoplasmic reticulum of the hepatocytes leading to AAT deficiency (AATD). Z-AAT is the most severe and common deficient variant. This mutant is not only retained in the endoplasmic reticulum but accumulates as an aggregate that triggers a cascade of intracellular signalling pathways inducing hepatocyte injury and death. Nevertheless, among all the homozygous ZZ patients only 15% develop liver injury, with a wide-range of disease severities ranging from hepatic fibrosis to cirrhosis or even hepatocellular carcinoma. Due to the lack of knowledge surrounding modifiers associated with Z-AAT-mediated hepatocyte toxicity, it is impossible to screen for AATD patients at risk of liver damage and to develop accurate therapeutic strategies. This review aims to give an overview and update our knowledge of AATD associated with liver disease and discusses possible new therapeutic strategies.
INTRODUCTION
Alpha-1 antitrypsin deficiency (AATD), described in 1963 by Laurell and Eriksson,1 is a rare inherited disorder with a prevalence of approximately 1 in 2,000–5,000 births in North American and European populations. AATD is associated with lung (emphysema) and/or liver damage (cirrhosis). Mutations in SERPINA1 (chromosome 14), which encodes Alpha-1 antitrypsin (AAT) protein, cause AATD, which leads to a reduced level of AAT in serum.2,3 The secretion rate functionality of AAT depends on the inherited mutation.3
Hepatocytes are the major source of AAT production in the liver. Around 1.5 g/L of this protein is secreted into the bloodstream in healthy adults. The main function of AAT is to protect the lung from non-specific protease-mediated degradation. AAT is carried by blood to the lung interstitium and the alveolar lining fluid, where it inhibits neutrophil serine proteases, such as neutrophil elastase (NE), cathepsin G, and proteinase-3. AAT is the main natural antagonist of NE. The latter is responsible for elastin degradation, the main constituent of the alveoli essential for the lung function.4 NE also presides over a wide range of pro-inflammatory actions potentially leading to several types of lung alterations, including emphysema and bronchiectasis.5 Thus, a reduced AAT serum concentration, as observed in AATD, results in lung tissue damage and major respiratory diseases, such as emphysema.6
In addition to the lung issues, some AATD patients may also develop liver diseases such as cirrhosis or hepatocellular carcinoma (HCC). AATD is the most common genetic cause of liver disease in children.7 Over the past 20 years, there has been an increase in the prevalence of the adult form of AATD-mediated liver disease. More than 88% of patients who undergo liver transplantation for AATD are adults, with the peak age range being 50–64 years. The adult form of the disease seems to be an age-dependent degenerative disease. This is in contrast to the paediatric form of the disease, the progression and severity of which is associated with other genetic factors.8
This wide variation in the severity and form of liver disease among patients is still not well understood, but genetic predisposition could play a role.9,10 However, the genetic underpinnings of AATD remain unclear, and new mutations, including severe deficient mutations, are yet to be detected. A better understanding of the genetic factors involved in AATD-mediated liver disease could help in the development of new therapeutic strategies. This review aims to provide an overview of our understanding of AATD-mediated liver disease, from the clinical features to the molecular mechanisms. Additionally, it opens a discussion about the therapeutic prospects.
GENERALITY: ALPHA-1 ANTITRYPSIN PRODUCTION, SECRETION, AND ACTION
SERPINA1 is highly polymorphic, with more than 100 alleles identified.11 The gene is composed of 7 exons and 6 introns located on the q arm of the human chromosome 14 (14q32.1). Transcription of SERPINA1 results in 11 mRNA divided into 5 mRNA products: 1.8, 1.9, 1.95, and 2.0 kb-sized transcripts expressed by monocytes, and a 1.6 kb-sized transcript expressed exclusively by hepatocytes. They are generated by variability in transcription start sites and alternative splicing of untranslated exons according to tissues. A translation site beginning at exon 2 results in the production of 418 residues with a 24-residue peptide signal.12 This peptide signal promotes the targeting of the AAT protein towards the endoplasmic reticulum (ER). In this organelle, AAT is 3-N glycosylated and then acquires native form in the Golgi. AAT is composed of two ß sheets, nine α helices, and a reactive loop, and is secreted into the bloodstream.
AAT is the most abundant serine proteinase inhibitor in human plasma and is mainly produced and secreted by hepatocytes. However, other cells such as neutrophils, macrophages, monocytes, and epithelial cells also produce AAT in smaller quantities. AAT plasma concentration varies from 0.9–2.0 g/L and according to the body’s inflammatory regulation.13 During inflammation (acute phase), AAT levels increase rapidly by 3–4-fold. The protein inhibits a range of pro-inflammatory proteases, such as proteinase 3, cathepsin G, and NE.14 The mechanism of inhibition has been determined by crystallographic studies. Targets, such as NE, bind to the reactive loop of AAT, which constitutes a trap for the target protease, and cleave at a precise site causing a conformational transition. The amino terminal half of the reactive loop is inserted into the main ß-sheet, which acquires an extra strand. The reactive loop residue, located upstream of the cleavage, flips from the upper to the lower pole of the protein, carrying with it the protease trapped within a covalent complex. The resulting inactive AAT/protease complex is highly stable.15
AAT is crucial in inhibiting these molecules and maintaining the balance of protease/anti-protease that are important for lung integrity. Consequently, AATD predisposes patients to lung injury.
ALPHA-1 ANTITRYPSIN DEFICIENCY
As previously mentioned, AATD is caused by a mutation in the SERPINA1 gene that predisposes not only to lung injury but also liver damage. Over 100 mutations have been described to date and are classified as deficient or null.16 Null variants are characterised by undetectable levels of AAT in the serum due to nonsense mutations or frameshifts leading to a premature stop codon. However, deficient variants, such as the well-known Z-variant, are characterised by low levels of circulating AAT, generally due to a point mutation or small deletions.16
The two variants named ‘Z’ and ‘S’ are the most common. These mutants are so named because of their isoelectrofocussing pattern in which they migrate slowly compared to the wild type (WT) isoform referred to as M-AAT. These two variants are caused by point mutations (Glu342Lys and Glu264Val, respectively), which result in aberrantly folded protein.1,17 The Z variant is the most severe form and its mutation results in the aggregation of Z-AAT, resulting in greater plasma deficiency and potentially hepatocyte toxicity and liver damage.
Z-AAT Mutant
As previously mentioned, the Z-variant is the result of a lysine substitution by glutamate at position 342. This mutation leads to the retention of the Z-variant in the ER,18 where it interacts with chaperones such as Grp78/BiP, Grp94, Grp170, and calnexin, all of which have been shown to contribute to its ER retention (Figure 1).19
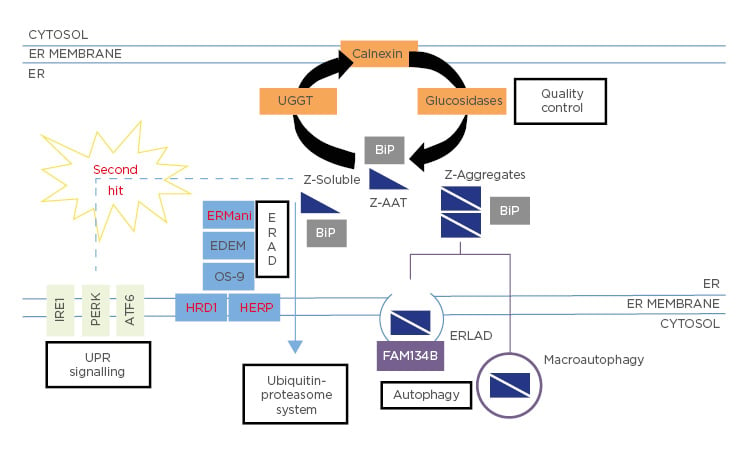
Figure 1: Fate of Z-Alpha-1 antitrypsin within the endoplasmic reticulum.
The Z-variant is retained within the ER in both soluble and aggregate forms. The latter may be bound into the hepatocytes in inclusion bodies (IB) and may predispose to liver injury.20
This histopathological hallmark of the disease stains positively in hepatocytes with periodic acid–Schiff after treatment with diastase. The mechanism of IB formation is not clearly understood. It was proposed that these IB are a part of the ER components and could have a protective effect on hepatocytes.21 Moreover, there is heterogeneity in the distribution and size of IB within hepatocytes, many of which are free or resemble small clouds of ‘dust’, while others are huge and well-marked. This difference and its association with the disease outcome is yet to be explained.
These structures are formed by the opening of ß-sheet, which can accept the loop of another molecule to form a dimer that extends into aggregates.18 Due to the retention of the Z mutant in the ER of hepatocytes, homozygous ZZ patients have approximately 85% lower circulating levels of AAT. The retention and accumulation of this mutant in the ER could have a gain-of-function toxic effect leading to hepatocyte toxicity and death, thus predisposing homozygotes to liver diseases.22
Other Mutant-Mediated Liver Disease
The Z-variant is not the only AAT mutant involved in liver diseases. Rare variants, such as Mmalton (Phe51/52 del) and Siiyama (Ser53Phe), may cause liver disease (Table 1).23,24
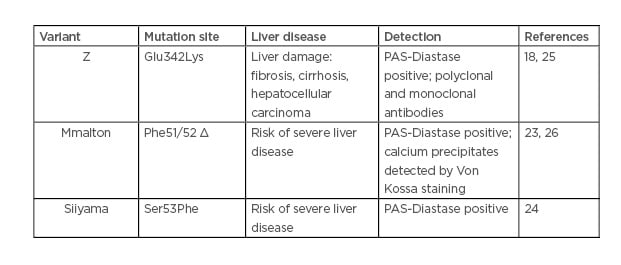
Table 1: Alpha-1 antitrypsin variant-mediated liver disease.
An Italian study has reported that among subjects with severe AATD, 11% carried a genotype other than the common Z or S/Z, and 13% of this population had developed a chronic liver disease.27 According to the Spanish Register of AATD patients, Mmalton is the most prevalent deficient rare variant.28 Unfortunately, given the extreme rarity of such variants, their detection seems to be underestimated.
Similar to the Z mutant, Mmalton and Siiyama mutants form polymers and IB in the hepatocytes. However, the formative processes and nature of these aggregates are different depending on the variant. First, only the Z aggregates are recognised by the non-commercial monoclonal antibody ATZ11.25 In addition, IB resulting from the Mmalton variant contain calcium precipitates.26 A better diagnostic, molecular characterisation, and identification of the factors and stress pathways induced by these rare variants must be undertaken.
ALPHA-1 ANTITRYPSIN DEFICIENCY AND LIVER DISEASE
In 1969, Sharp et al.29 were the first to establish a link between AATD paediatric homozygous ZZ patients and liver disease. It is now well-known that AATD may be associated with liver damage in infancy, childhood, and adolescence, as well as later in adult life.
However, not all patients develop liver disease, and the symptoms and outcomes of the disease are highly variable: these include the age or stage of the first manifestation, as well as the rate of progression of the damage. Both an acute or chronic form of liver injury may be encountered. A recent review of the Organ Procurement and Transplantation Network database for liver transplantation in the USA has shown that there are two peak ages for severe AATD-associated liver disease: 0–5 and 50–65 years of age.8
In brief, a small subset of affected children will develop severe liver disease in the first decade of their life, others may develop and grow up normally, because most adolescents display normal liver enzyme levels30 even in the presence of advanced liver disease.31
In this chapter, the authors discuss the clinical characteristics of the disease, such as the different forms and symptoms associated with AATD during childhood and adulthood. We also discuss the incidence of genetic and environmental factors and the heterozygous status in the development of liver diseases associated with AATD.
Liver Disease During Childhood
The main clinical characteristic of childhood liver disease is usually persistent jaundice, which is a general hallmark of neonatal cholestasis observed during the first 1–2 months after birth. In addition to this, infants may present with itching, bleeding, trouble eating, and elevated liver enzyme levels. Although neonatal cholestasis disappears in 3–6 months, in severe cases, this cholestasis may progress to cirrhosis with early development of portal hypertension and liver failure.32 In toddlers, the liver damage associated with AATD may present without chronic liver disease or with isolated signs such as an accumulation of fluid in the abdominal cavity (ascites), splenomegaly, or hepatomegaly.
Much of what is known about AATD-associated liver disease during neonatal period and during childhood comes from a prospective study by Sveger,33 who has determined the frequency of liver disease. In his study, Sveger33 screened 200,000 newborns in Sweden and identified 127 homozygous ZZ patients who were monitored until 12 and 18 years of age. From this group of homozygotes, 14 had prolonged obstructive jaundice and 9 of the 14 had severe liver disease. The authors concluded that only 15% of homozygous ZZ patients had developed clinically significant liver disease in the first 4 decades of life, and <3% of those had progressed to life-threatening end-stage disease as infants.33,34 Recently, a study from the French paediatric cohort named DEFI-ALPHA has observed that in France, 18.3% paediatric AATD patients had a severe liver disease and almost half of them ended up with a liver transplantation.35 The lower rate of liver disease reported in the Swedish study might be due to the mode of patient recruitment. Indeed, the patients in the study by Sveger33 were recruited through neonatal screening, whereas only patients with known AATD were included in the French study.
As previously mentioned, the majority of AATD patients are healthy or free of chronic disease by 18 years of age. However, for some of them, this period of few or no signs of liver damage will end and severe, progressive liver disease will onset during their adulthood.
Liver Disease During Adulthood
AATD adults may develop chronic hepatitis, cirrhosis, and HCC, and the incidence of liver disease increases with age.36 Liver disease is more common in adults than in children. Chu et al.8 have shown that currently more than 88% of patients who undergo a liver transplantation for AATD are adults, with the peak age range being 50–64 years. This condition is under-recognised and undiagnosed during adulthood because biochemical and histopathologic analyses in homozygous ZZ adults may produce results similar to those for alcoholic liver disease.
In adult AATD, advanced liver fibrosis is present without clinical liver disease, and fibrosis on biopsy is detected in approximately one-third of AATD adults.37-39 In addition, several studies have reported obesity as a risk factor for liver disease in AATD adults.39,40 The prevalence of hepatic steatosis in adult AATD is higher than 20–30% expected for the general USA population and represents a secondary cause of hepatic steatosis similar to other genetic/metabolic diseases. Valenti et al.41 evaluated the impact of the two most relevant AAT variants in two large cohorts of nonalcoholic fatty liver disease and chronic alcohol misuse. The authors found that the Z-variant is a major risk factor for cirrhosis in the context of chronic metabolic injury such as nonalcoholic fatty liver disease and chronic alcohol misuse.41
To summarise, risk factors for adults with liver disease in AATD are age >50 years, male sex, repeated elevated liver enzymes, hepatitis virus infection, obesity, diabetes, and metabolic syndromes.39
Liver Disease and Genetic and Environmental Influences
As previously mentioned, only about 15% of homozygous ZZ patients develop clinically significant liver disease in their childhood. Other co-factors, such as hepatitis, obesity, metabolic syndromes, or alcohol intake, could augment the risk of hepatic disease during adulthood but obviously not during childhood. Consequently, the modifiers in the infant form of severe and progressive liver disease remain unidentified. Since not all patients with the homozygous ZZ genotype develop end-stage disease, it has been suggested that genetic and environmental factors may be implicated in variability of onset and disease severity. From a clinical point of view, AATD patients with neonatal cholestasis are likely to develop severe liver disease.35 Other risk factors, such as male sex or the absence of breastfeeding, were found to not be associated with severe liver damage.35 From a cellular/genetic perspective, in 2009, based on a candidate gene-sequencing strategy, Pan et al.10 demonstrated that differences in ER mannosidase I expression were associated with an earlier age-of-onset for end-stage liver disease. However, the significance of this association has been challenged, as a replicate study in another cohort failed to reproduce the results.42 Recently, a study that focussed on the MAN1B1 gene (that encodes the ER mannosidase I protein) and SERPINA1 gene showed that no genetic polymorphisms in these two genes influence the onset and severity of liver disease in AATD during childhood.43 Finally, the authors’ group had identified two mutations, HERPUD1 R50H and HFE H63D, that were associated with the advanced liver disease component of AATD.9 Based on this work, they have also observed that specific pathways, including ER-associated degradation pathway (ERAD) and Unfolded Protein Response (UPR), could be novel risk factors for AATD-caused liver disease (Figure 1).9 However, these results are in line with the findings of a large-scale screening study for AATD.9
The AATD-associated liver disease during adulthood could be predominantly an age-dependent degenerative disease. This hypothesis is consistent with different studies using an AATD mice model that showed differences in the activation and expression of proteins and molecular pathways between livers from young mice and older mice.44,45 These results suggest that different modifiers (co-factors, stress pathways) are involved in the infant and the adult form of the disease, and that their identification is crucial for accurate treatment of patients.
Alpha-1 Antitrypsin Deficiency and Hepatocellular Carcinoma
Although the incidence of AATD on HCC development is still controversial, some studies have pinpointed AATD as a risk factor for the development of the cancer.46 Based on the AATD mouse model,47 the livers of 79 Z-AATD and 18 WT mice were analysed. Liver pathology was seen more frequently in Z-AATD livers (47/79) than in WT (5/18), a development that was also age-related. In older Z-AATD mice (18–24 months), livers showed malignant tumours (HCC and angiosarcoma) (17/50), hyperplastic nodules (28/50), and non-specific changes (33/50), whereas only 9/50 were normal.47 Similarly, a Swedish autopsy study based on 31 autopsied adults with severe AATD has shown that in the homozygous ZZ group, there were 13 cases of cirrhosis, 5 cases of HCC, and 8 cases of gallstone disease.48 Consequently, AATD patients are at greater risk of cirrhosis and HCC, with the risk of HCC being higher in males.
From a molecular perspective, a comparative ‘omics’ approach between diseased human-induced pluripotent stem cell-derived and WT hepatocytes has pinpointed specific proteins associated with predisposition to malignancy that are highly upregulated in the former.49 More recently, another study has examined the contribution of DNA methylation to AATD adult liver disease heterogeneity, where the global analysis revealed significant genomic hypomethylation in AATD liver impacting genes related to liver cancer.50
Heterozygotes
Retention of Z-AAT within hepatocytes is responsible for liver disease. Recently, a clear relationship between Z-AAT accumulation and fibrosis was demonstrated.39 According to that study, only patients carrying aggregate-forming AAT variants are susceptible to developing liver disease. Thus, it is not surprising that heterozygous patients, such as those with SZ or MZ variants, can also develop severe liver disease in childhood and adulthood.35,51 Hence, it has been suggested that heterozygosis increases the risk of developing liver disease. Interestingly, the incidence of liver disease could be higher in heterozygotes with the deficiency than in the general population, especially if the affected individuals have other liver comorbidities.5 Many patients who undergo liver transplantation with a diagnosis of AATD are actually heterozygotes who also have other risk factors (e.g., alcohol usage or steatosis).8,52 Recently, two cohort studies have shown that that Z allele is a risk factor for cirrhosis in alcoholic and nonalcoholic fatty liver diseases.53 Among patients with cirrhosis, decompensation of cirrhosis with ascites or encephalopathy was significantly more frequent in patients with MZ than in homozygous MM or WT allele patients.54 The MZ genotype is a genetic risk factor for more advanced cirrhosis and decompensation, especially if the affected individuals have other liver comorbidities. Z heterozygous variants seem to be strong risk factors for the severity of chronic liver diseases.
Given that the MZ genotype represents 2% of the USA and European populations, and the fact that the Z allele represents a strong disease modifier, Z allele carriers are at higher risk. Hence, genotyping for the Z allele should be considered as the first-line test in all patients with cirrhosis. Finally, further research is needed to define the clinical features of heterozygous Z and S carriers and determine whether those AAT variants present a clinical phenotype.53,54
In conclusion, there is a wide variation in the forms and severity of liver injury among AATD patients and, as mentioned above, very little is known about predispositions. This variability may be related to how the affected individual responds to the intracellular accumulation of Z-AAT in hepatocytes.
Z MUTANT AND MOLECULAR MECHANISM: FROM DEGRADATION TO DEGRADATION TO HEPATOCYTE TOXICITY
Z-Variant Disposal
In the ER, Z-AAT mutant accumulates both in soluble and aggregate forms (Figure 1). Several lines of evidence suggest a dose–response relationship between Z-AAT intracellular protein accumulation and liver injury. Thus, the accumulation of these aggregates is the first event of liver injury cascade. To prevent this dramatic issue, hepatocytes use different degradation pathways to protect themselves from proteotoxicity. Depending on the form of the mutant, soluble or aggregated, different degradation pathways have been identified (Figure 1). The soluble form, which represents most of the intracellular forms of the Z-variant (>80%), interacts with several proteins involved in important pathways, such as quality control or ERAD, before finally being degraded by the proteasome (Figure 1). Previous work has shown that among all the molecules involved in those pathways, one molecule present in the ER, calnexin, is a critical point of control. Calnexin is a transmembrane ER chaperone that binds Z-AAT and targets it for degradation. For instance, it has been shown that inhibitors indirectly affecting the interaction of the Z mutant with calnexin, such as kifunensin (mannosidase inhibitor I and II) or castanospermine (glucosidase inhibitor), restore the secretion of this variant.55 In addition to calnexin, studies in human fibroblast cell lines from homozygous ZZ patients have shown that patients with liver disease have less efficient Z-AAT degradation than homozygous ZZ patients without liver disease.56 These results suggest that many other proteins involved in the disposal of the Z mutant could be critical in the liver damage linked to AATD. Among all these potential proteins, different studies have pinpointed the ERAD pathway, with genetic modification in genes encoding the proteins Man1B1 or Herpud1 perhaps altering susceptibility to liver injury by changing the efficiency of Z-AAT degradation.9,35
The autophagy pathway is another degradation pathway activated by the cell to prevent liver toxicity. This is a highly conserved and important degradation system specialised in disposal of protein aggregates and large structures via the formation of autophagosomes. Consequently, autophagy could rather be involved in the degradation of Z mutant aggregates that are not capable of being eliminated by the protea some. It has been shown that autophagosomes were abundant in the hepatocytes of homozygous ZZ patients and that Z-AAT retention in the ER is associated with a marked autophagic response.50 Consequently, some studies have observed that enhancing autophagy may reduce hepatotoxic effect. For instance, weekly administration of rapamycin, a mTOR pathway inhibitor, in a mouse model increased autophagy and diminished Z-AAT aggregates accumulation, subsequently leading to reduction of liver injury.57 This is also observed using another autophagic drug, carbamazepine (CBZ), a drug that reduces the hepatic fibrosis and inflammatory response associated with the Z-AAT aggregates in a mouse model.58
Nevertheless, several questions concerning autophagy and AATD are still unclear and controversial. What activates this pathway? Which type of autophagy (e.g., ER-phagy, macroautophagy) is activated by the aggregates? What is the mechanism of vesicle/autophagosome formation? Some studies suggest an intervention of macroautophagy in the clearance of luminal aggregates; this would imply their dislocation across the ER membrane, or the capture of ER portions containing them by autophagosomes.59 Conversely, other studies based on the transcription factor EB master gene (TFEB) that regulates the autophagy pathway have speculated that mechanisms independent of mTOR or other classical macroautophagy were involved in the disposal of Z-AAT aggregates.58,60 In this case, autophagy is induced due to reduced intracellular calcium and inositol levels. Finally, it has been recently described that an ER-to-lysosome-associated degradation pathway was implicated in the degradation of Z-AAT aggregates.61 This pathway involves calnexin and the engagement of the LC3 lipidation machinery by the ER-resident ER-phagy receptor, FAM134B (Figure 1). Consequently, Z-AAT aggregate delivery from the ER lumen to endolysosomes for clearance does not require ER capture within autophagosomes. Rather, it relies on vesicular transport, where single-membrane, ER-derived, Z-AAT-containing vesicles release their luminal content within endolysosomes upon membrane-membrane fusion events.61
Among these two degradation pathways, ERAD/proteasome and autophagy, it remains unclear whether autophagy is a specific response to the accumulation of Z-AAT or rather a secondary process that becomes more important when ERAD and/or the proteasome are overwhelmed. Based on a yeast Z-AAT expression system, it seems that the trigger for induction of autophagy by Z-AAT might be the formation of Z-AAT aggregates.62 Indeed, at low levels, the Z mutant remains soluble and is disposed by the ERAD/proteasome pathway. Conversely, higher levels of Z-AAT expression induce aggregate formation, thereby activating the autophagy pathway required for their degradation. Another study on Z mouse liver extracts showed that polyubiquitin conjugates were accumulating, despite normal recruitment to catalytically active 26S proteasomes. This suggests that a defect at the 26S proteasome, other than the compromised binding to polyubiquitin chain or peptidase activity, plays a role in this accumulation.63 It is still unknown whether this event reflects a lack of response to an increased demand for the proteasome or a response counteract by rapid elimination of damaged subunits and/or complexes.
In addition to these two major degradation pathways, the cell can also protect itself from Z-AAT aggregates by activating other degradation pathways. For example, in a murine hepatoma cell line, Z-AAT is degraded by a non-proteasomal mechanism, which is sensitive to tyrosine phosphatase inhibitors.64
Even if hepatocytes use different systems to protect themselves from the Z-AAT proteotoxicity consequences, in 15% of the homozygous ZZ patients those degradation pathways are not sufficient and Z-AAT aggregates will trigger multiple signalling events, finally leading to cellular toxicity and death.
Z Mutant and Proteotoxicity
Although several aspects of the disease pathogenesis are still unclear, accumulation of Z-AAT aggregates can affect various intracellular signalling pathways leading to injury cascade, including apoptosis. Hence, mitochondria have been closely associated with the Z-AAT aggregates toxicity via their role in apoptosis induction. In a Z-AAT mouse model, both mitochondrial and liver injuries were reduced with the administration of cyclosporin A, an inhibitor of mitochondrial permeability transition.65 Recently, transcriptome and proteome analyses using human induced pluripotent stem cells derived from patients with Z-AAT differentiated into hepatocyte-like cells in comparison to patient-specific genetically corrected hepatocyte-like cells, have confirmed that Z-AAT aggregates were associated with disrupted mitochondrial structure.49
Other pathways have been linked to the Z-variant and cell toxicity. For instance, the accumulation of Z-AAT aggregates triggers NF-κB signalling through a pathway named ‘ER overload response’.66 Although this response remains unclear, it seems that a leakage of calcium from the ER might have a role in its activation. Moreover, it has been shown that the kinases c-Jun N-terminal kinase 1 (JNK1), 2 (JNK2), and c-Jun play important roles in the pathogenesis of the liver disease by controlling the degree of Z-AAT accumulation.67 Finally, oxidative stress was shown to contribute to liver damage in a murine model of Z-AAT. Higher levels of reactive oxygen species and a more oxidised cellular redox state are observed in liver tissue from Z-AAT mice compared to WT mice.67
Interestingly, the accumulation of intracellular Z aggregates is associated with multiple pathways, but in the absence of the induction of the UPR. This pathway is generally activated when misfolded proteins accumulate within the ER. Thus, UPR activation reduces the entry of nascent proteins into the ER and improves the folding and degradation of misfolded proteins. One hypothesis is that the absence of UPR activation might be caused by the structure of the Z-AAT aggregates. Indeed, these structures are relatively well-folded, and they might not contain hydrophobic areas/misfolded parts. Nevertheless, the accumulation of Z-AAT aggregates seems to sensitise the cell to a ‘second hit’ that can induce an UPR more efficient than that observed in WT cells (Figure 1).9,68
THERAPEUTIC STRATEGIES
Currently, there is no specific treatment for AAT-associated liver disease, other than standard liver supportive care, such as ursodeoxycholic acid.69 Generally, management of the disease focusses on preventing the complications of chronic liver disease.69 In severe cases, when life-threatening liver disease does develop, liver transplantation is performed with excellent success rates.69 Following liver transplantation, the serum AAT levels return to normal.69 To overcome the absence of specific treatments, several therapeutic strategies have been employed. They consist of interfering in the different pathways in which the Z-variant is involved (Table 2).
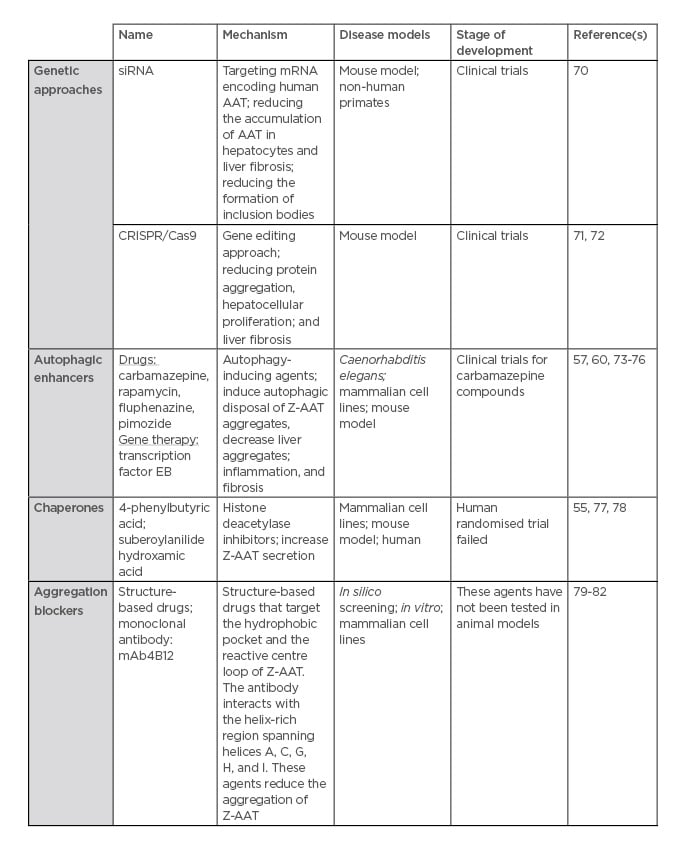
Table 2: Therapeutic strategies for alpha-1 antitrypsin deficiency-associated liver disease.
Reducing the Amount of Z-AAT Aggregates
Since the accumulation of the Z-variant aggregates into the ER can trigger liver damage, one of the first strategies implemented is to reduce this accumulation in the ER.
For this purpose, small interfering RNA (siRNA), targeting mRNA encoding human AAT, have been developed by two biotechnology companies: Arrowhead Research Corporation and Alnylam Pharmaceuticals, Inc. In transgenic mice expressing the Z-variant, siRNA treatment reduced the accumulation of Z-AAT in hepatocytes, the formation of inclusion bodies, and liver fibrosis.70 Moreover, in non-human primates, 80% of AAT in the bloodstream is reduced following siRNA treatment. This strategy has been extended to humans. In June 2015, Phase I/II clinical trials using the siRNA named ALN-AAT was conducted in healthy patients and homozygous ZZ patients who developed liver damage. Although circulating AAT levels decreased significantly (about 75% on average) up to 6 months at a single dose of 6 mg/kg, liver enzymes increased in three patients, indicating liver.83 The other candidate is the Arrowhead siRNA AAT, named ARC-AAT. A Phase I study, in healthy volunteers and AATD patients, had been launched and was terminated in January 2017. ARC-AAT was well-tolerated and induced deep and durable reduction of AAT. Thus, a Phase II study was recently initiated to determine the safety and effect on circulating and intrahepatic AAT levels of ARC-AAT.84,85 Further details about and the conclusions of this programme are pending.
Rather than targeting mRNA, another way to prevent the accumulation of Z-AAT in the ER would be to directly correct the mutated gene by using a gene editing approach, such as CRISPR/Cas9. The CRISPR/Cas9 approach uses a guide RNA, which is homologous to that of the DNA to be excised/targeted, and a Cas9 enzyme, which is an endonuclease, the co-operation of which leads to a DNA double strand break at a specific location. This approach has already been used in a transgenic mouse model expressing human Z-AAT.71 A guide RNA specific for hSERPINA1 expressed in the liver has been used with the aim of disrupting the gene and reversing the disease phenotype. Thus, with this treatment, the author has shown a reduction in protein aggregation, hepatocellular proliferation, and liver fibrosis without off-target DNA editing.72
Increasing the Degradation Pathway
Autophagy and the ERAD/proteasome pathway mainly remove the aggregate and the soluble Z-AAT forms, respectively. Therefore, one of the strategies would be to increase these degradation pathways. The most promising advances have been with autophagy enhancer treatments. One interesting avenue involves CBZ, a drug largely used for epileptic treatment, which has been shown to enhance both autophagy and proteasome pathways in AATD cellular and mouse models. CBZ increases intracellular degradation of the Z-variant and, in a mouse model, administration of large doses (>10-times the recommended dose for humans) reduced intrahepatic Z-AAT inclusion and reversed hepatic fibrosis associated with AATD.58 Following these encouraging results in 2012, a Phase II clinical trial has been conducted on 30 AATD patients with severe liver disease. This clinical trial will be finished in 2020 and will provide us more details on CBZ efficacy in humans.86
As observed with CBZ, another pro-autophagic agent, rapamycin, has been shown to be effective in clearing Z-AAT aggregates in cell lines and in mice that express the Z-variant. This was associated with a reduction in markers of hepatocellular injury, such as hepatic fibrosis.57 Given these findings, and as these two pro-autophagic agents do not act through the same molecular pathway, a combined treatment of CBZ (mTOR-independent) and rapamycin (mTOR-dependent), would be interesting to test. In the light of pro-autophagy and AATD, a drug screen based on an AATD Caenorhabditis elegans model has identified several other drugs that enhance the autophagic disposal of Z-AAT.73 Finally, an alternative approach to enhance autophagy is to use a viral vector that expresses the transcription factor TFEB, which is a master regulator of autophagy. TFEB localises to the lysosomal membrane, and, upon dephosphorylation, the factor is activated and transported into the nucleus where it acts as a transcription factor. TFEB has been shown to modulate lysosomal clearance74 and induce autophagy.75 The effect of TFEB gene transfer on the induction of autophagy and lysosomal biogenesis has already shown efficacy in lysosomal storage diseases.76 Concerning AATD, it has been shown in a mouse model that gene transfer of TFEB reduced the accumulation of Z-AAT, liver apoptosis, and fibrosis.60
Improve the Folding/Inhibit Polymerisation
Another strategy to reduce the accumulation of Z-AAT is to improve its folding and/or increase its secretion by, for instance, using chemical chaperones that can facilitate mutant protein folding and trafficking. Among these chemical chaperones, suberoylanilide hydroxamic and 4-phenylbutyrate, two histone deacetylase inhibitors, have been shown to increase the secretion of Z-AAT in cellular and in vivo models.55,77 However, a human randomised trial failed to demonstrate the efficacy of 4-phenylbutyrate.78
Another avenue of treatment could be a ‘structure-based’ drug development.79 A better knowledge of polymer formation has permitted drug discovery. Polymers have a hydrophobic pocket, which accepts an exogenous reactive loop peptide. Mutation in this hydrophobic pocket has been initially shown to reduce Z-AAT aggregate formation and increase the secretion of Z-AAT using a Xenopus oocyte expression system.80 Furthermore, small-molecule drugs that target the hydrophobic pocket and peptides that target the reactive centre loop of AAT have been tested and were found to prevent Z-AAT aggregation.81 Nevertheless, some of these drugs and peptides increase intracellular degradation rather than Z-AAT secretion, or improve the rate of secretion but lead to more Z-AAT intracellular accumulation. Moreover, these peptides have not been tested in animal models.
Recently, monoclonal antibodies such as mAb4B12 were shown to reduce the accumulation of Z-AAT polymers in vitro but failed to prevent Z aggregation and increased its secretion in cellulo.82
Other Treatments
As mentioned previously, oxidative stress has been associated with AATD liver damage. Antioxidant therapy, such as with Vitamins A, C, and E, and N-acetylcysteine could be interesting to test and evaluate.
With the recent advances in our knowledge of gene editing, a cell transplantation therapy for AATD could be also considered. Ding et al.87 demonstrated that WT donor hepatocytes could almost completely repopulate the liver of the AATD mouse model. In addition, a biallelic correction of the Z-AAT point mutation (Glu342Lys) in human iPSC (from a homozygous ZZ patient) had been achieved by a combination of zinc finger nucleases and piggyBac technology.51 Subsequently, these cells were engrafted into the liver of a transgenic mouse model, which were able to colonise the liver in vivo and display functional activities. This exciting strategy could protect against COPD and liver damage associated with AATD.
CONCLUSION
AATD is a genetic disorder associated with an increased risk of liver disease in children and adults. AATD is caused by mutations in the SERPINA1 gene encoding the AAT protein. Among mutations responsible for AATD, Z mutant is the most severe and common deficient variant. Homozygous ZZ patients have low circulating level of AAT. This is caused by the retention and accumulation of soluble and aggregate Z-variants in the ER of hepatocytes where it is mainly synthesised and secreted. The accumulation of Z-variant aggregate triggers intracellular signalling pathways, such as apoptosis, NF-κB, and JNK signalling, and causes liver damage. Some homozygous ZZ patients may develop liver damage, with variable severity, such as cirrhosis or HCC. The cause of this variability in severity of liver disease remains unclear. The development of new therapies, for example RNA technology, gene repair (CRISPR/Cas9), stimulating degradation pathways, and inhibiting aggregation via chemical chaperones and drugs, would negate the need for liver transplantation, which is currently the only curative treatment.
In the future, one of the major challenges will be to identify modifier-mediated Z-AAT toxicity and understand how these modifiers act. This will open a new personalised approach to treatment and could be used as biomarkers of the hepatic disease outcome. Moreover, further studies are needed on liver disease among adults with AATD and on the potential risk to subjects carrying rare variants of the disease.






