Abstract
Non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH), which are usually associated with obesity and metabolic syndrome, are considerable health and economic issues due to the rapid increase of their prevalence in Western society. Histologically, the diseases are characterised by steatosis, hepatic inflammation, and if further progressed, fibrosis. Dietary-induced mouse models are widely used in investigations of the development and progression of NAFLD and NASH; these models attempt to mimic the histological and metabolic features of the human diseases. However, the majority of dietary mouse models fail to reflect the whole pathophysiological spectrum of NAFLD and NASH. Some models exhibit histological features similar to those seen in humans while lacking the metabolic context, while others resemble the metabolic conditions leading to NAFLD in humans but fail to mimic the whole histological spectrum, including progression from steatosis to liver fibrosis, and thus fail to mimic NASH. This review summarises the advantages and disadvantages of the different dietary-induced mouse models of NAFLD and NASH, with a focus on the genetic background of several commonly used wild-type mouse strains as well as gender and age, which influence the development and progression of these liver diseases.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) have many medical and economic implications worldwide and in all age groups, confirmed by clinical studies which indicate an almost 2-fold increase in mortality for NAFLD patients.1-11 NAFLD and NASH are closely associated with obesity, insulin resistance, and glucose intolerance, and further represent the hepatic manifestation of metabolic syndrome (MS).1,2,12-14 The diseases include different stages and display a broad spectrum of hepatic pathological features, ranging from mild steatosis to significant inflammation, fibrosis, and finally, cirrhosis.13,15-17 Additionally, in humans NAFLD and NASH are histologically characterised by hepatocellular degenerations like ballooning and the formation of Mallory–Denk bodies.18
Experimental studies regarding NAFLD/NASH often use mouse models, due to their similarity to human anatomy, genetics, and physiology. Many aspects of the diseases can be studied in a cost and time-effective manner (e.g. body composition can be monitored, metabolic parameters can be assessed, and pathohistological alterations can be induced within a relatively short time).19-30 Nevertheless, mouse models are restricted by the available sample amount and the difficulties of invasive or surgical procedures (such as in haemodynamic studies) in mice due to their size.31,32 For modelling NAFLD/NASH in mice, several different wild-type (WT) strains (e.g. C57BL/6, Balb/c, and 129Sv) fed with special diets (e.g. high-fat [HF], methionine and choline-deficient [MCD], and high-fructose diets) have been previously used.22-30 These dietary mouse models attempt to mimic hepatic and metabolic conditions occurring during human NAFLD/NASH. Depending on the experimental setup, the models comprise distinctive features of human NAFLD/NASH, including hepatic injury (e.g. steatosis, lobular inflammation, ballooning, and perivenular fibrosis) and metabolic abnormalities (e.g. Type 2 diabetes and increased triglyceride and cholesterol levels). Furthermore, genetically modified mice (e.g. leptin-/- and acetyl-CoA oxidase-/- mice) are used to model human NAFLD and NASH.24,33-35 Thus, various mouse models are already established for investigating the complex pathophysiological mechanisms and influential factors that contribute to human NAFLD/NASH development, though the approaches, along with the results, vary greatly depending on the research question.5,36 The use of different genetic backgrounds for dietary and genetic mouse models is another variable, profoundly influencing outcomes of the respective studies. This review will summarise the phenotypes of dietary-induced mouse models of NAFLD and NASH and assess the influence of genetic background, gender, and age on the manifestation of these diseases in the respective mouse models.
DIETARY-INDUCED NON-ALCOHOLIC FATTY LIVER DISEASE AND NON-ALCOHOLIC STEATOHEPATITIS IN MICE
Methionine and Choline-Deficient Model
The induction of NAFLD/NASH in mice by a MCD diet is based on impaired phosphatidylcholine synthesis and the subsequent reduction in very low density lipoprotein (VLDL) production, leading to hepatic triglyceride accumulation and thus steatosis (Table 1).37 Furthermore, methionine and choline restriction promotes oxidative stress through the induction of hepatocyte microsomal cytochrome P450 2E1 (CYP2E1) expression and thereby increases reactive oxygen species formation.38,39
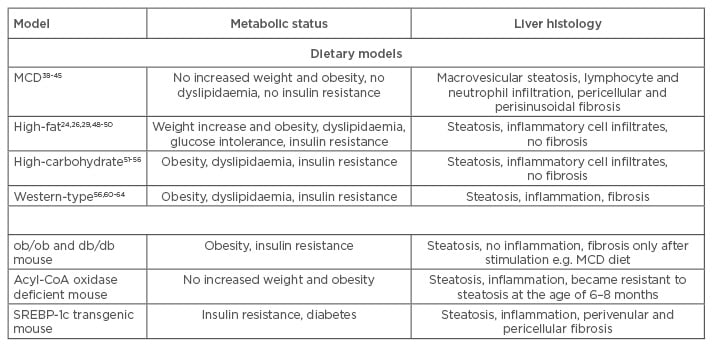
Table 1: Comparison of metabolic status and liver histology of commonly used dietary and genetic mouse models.
MCD: methionine and choline-deficient; SREBP-1C: sterol regulatory element-binding protein-1c.
Mice fed with a MCD diet develop steatosis within 3 weeks of feeding, expanding to extensive macrovesicular steatosis, including hepatic lymphocyte and neutrophil infiltration, after 8–10 weeks. Pericellular and perisinusoidal fibrosis is also observed after the respective feeding duration.39,40 The inflammatory response is increased in mice fed with a MCD diet (Table 1); liver macrophages are activated by the transcription factor nuclear factor kappa B, leading to increases in tumour necrosis factor alpha, interleukin 6, and transforming growth factor levels.41,42 Moreover, expression of intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and macrophage chemotactic protein-1 is increased, promoting hepatic infiltration and activity of neutrophils and macrophages.43
Although the hepatic histological features and inflammatory response of MCD models reflect the conditions in human NAFLD/NASH, this model type does not resemble human metabolic physiology; serum triglyceride, cholesterol, insulin, glucose, and leptin levels are not increased and mice do not exhibit peripheral insulin resistance. Furthermore, mice lose up to 25% body weight due to methionine and choline restriction (Table 1).41,44-46 This problem can be overcome by combining methionine-defined (0.1%) and choline-deficient feeding with HF content, which results in immediate development of hepatic steatosis, inflammation, and fibrosis along with moderate weight loss.47 Additionally, it has been shown that development of NAFLD/NASH in MCD animal models is species and strain-dependent. When three different rat strains and C57BL/6 mice were subjected to a MCD diet for 4 weeks, all rat strains developed steatosis, but inflammation was rare and fibrosis absent. In contrast, C57BL/6 mice showed necroinflammation and some exhibited focal pericellular fibrosis, while steatosis was minor compared with rats.48 The MCD model is a widely used and well-established model for investigations concerning inflammatory and fibrotic events in NAFLD and NASH in short-term courses of treatment compared with HF feeding. However, it is questionable if this model resembles the pathophysiological mechanisms of human NAFLD/NASH, which are closely associated with those of MS.
Models of High-Fat Feeding
HF diets attempt to induce NAFLD/NASH-associated liver injury in the context of obesity and MS (Table 1). These diets are composed of 45–75% fat with variations in saturated/unsaturated fat and cholesterol content. However, a study in which mice were fed with HF diets, varying in saturation of the fatty acids (FAs) and in cholesterol content, showed that steatosis and hepatic inflammation are almost independent of saturation of dietary FAs, and that different dietary FAs are mainly stored as oleic acid in mouse livers.30 Furthermore, dietary cholesterol seems to play a more pivotal role in the development of hepatic inflammation and thus may promote progression from NAFLD to NASH in mice.30,49
Lieber et al.25 reported a liquid HF diet composed of 71% fat, 11% carbohydrates, and 18% protein. This diet was shown to induce steatosis and inflammation in combination with metabolic abnormalities in rats and mice. Though the liver histology closely resembled features of human NAFLD, no signs of fibrosis were observed within this model, making it ineligible for studies regarding the progression of NAFLD to NASH.25,50,51 Another study investigated HF diet-induced NAFLD and NASH in a longitudinal approach by feeding mice a diet containing 60% fat (enriched in saturated FAs), 20% carbohydrates, and 20% protein for up to 50 weeks. After 10 weeks of HF diet, feeding-induced obesity and hyperinsulinaemia were observed, with glucose intolerance occurring after 12 weeks. Steatohepatitis was observed after 19 weeks of the HF diet, demonstrating that metabolic abnormalities are induced prior to development of steatosis and hepatic inflammation in HF diet-fed mice. Although features of human NAFLD were seen in this model, fibrosis was also not observed, even after 50 weeks of treatment.27
The use of HF diet mouse models is considered advantageous as the models are associated with relatively low experimental requirements and do not include non-physiological procedures to induce increased levels of triglycerides, hepatic inflammatory cell infiltration, obesity, and insulin resistance. However, most HF diet mouse models do not include fibrosis and induced liver injury is relatively minor compared with that seen in the MCD model (Table 1). Furthermore, periods of dietary treatment to induce features of NAFLD are lengthy, and results may vary because of treatment duration and experimental setup.
Models of High-Carbohydrate Feeding
Increased carbohydrate content can also be used to induce steatosis in mice (Table 1). Diets with varying carbohydrate content, composed of 30–65% glucose or fructose are given for 8 weeks, with fructose acting as the most powerful sugar to induce NAFLD. These mice exhibit obesity, steatosis, and inflammation in the context of metabolic changes, e.g. insulin resistance, increased levels of total cholesterol and triglycerides, and elevated liver enzymes.52-57 An excessive fructose intake is thought to increase de novo lipogenesis and visceral adipose tissue formation, resulting in increased portal delivery of free FAs to the liver and thus in hepatic triglyceride accumulation and inflammation (Table 1).57-60
Most often, increased carbohydrate uptake is combined with HF and high-cholesterol feeding; these diets are termed Western-type diets. Western-type diets can lead to hepatic triglyceride accumulation and steatosis along with hepatic inflammation and fibrogenesis, representing conditions similar to those seen in human NASH (Table 1).29,57,61-64 Thus, a combination of HF and high-carbohydrate content in the respective diets may cause a synergistic effect, which induces hepatic and metabolic conditions that better resemble the features of human NAFLD and NASH.65 Nevertheless, these models produce conflicting results that are more dependent on species and formulation of the particular diet, implicating the need for a strict experimental setup to generate reproducible results.
Genetic Mouse Models
Many genetic models have been established to investigate the role of the respective genes in NAFLD/NASH development and all are associated with hepatic lipid accumulation, though they concern various different pathways. Frequently used genetic models include mice that exhibit a mutation in the leptin (ob/ob) or the leptin receptor (db/db) gene, resulting in leptin deficiency or resistance to the actions of leptin. Subsequently, hyperphagia, obesity, metabolic abnormalities, and the spontaneous development of NAFLD, but no liver fibrosis, emerge (Table 1).24,34,35,66,67 Another category of genetic models are those which affect β-oxidation of long-chain FAs and hepatic triglyceride export; the mice, lacking acyl-CoA oxidase as an example, exhibit severe steatosis and inflammation, but no features of MS.33 Overexpression of the transcription factor sterol regulatory element-binding protein-1c (SREBP-1c) in mice leads to dysregulation of adipocyte differentiation and thus to insulin resistance, diabetes, and hepatic triglyceride accumulation, but this model is limited by a lack of obesity and metabolic abnormalities.68
INFLUENCE OF GENETIC BACKGROUND ON NON-ALCOHOLIC FATTY LIVER DISEASE/NON-ALCOHOLIC STEATOHEPATITIS DEVELOPMENT IN WILD-TYPE MICE
For humans it is evident that genetic variations influence the development and progression of NAFLD and NASH. For mouse models of diet-induced NAFLD and NASH, it is known that genetic background has a substantial influence on the observed NAFLD/NASH-associated pathological features.69
It has been demonstrated that when fed a MCD diet, the widely used inbred mouse strain, C57BL/6, is more prone to developing NAFLD/NASH- associated liver injury (e.g. lipid accumulation, oxidative stress, and fibrosis) compared with the C3H/HeN strain.70 Moreover, it was shown that when fed an MCD diet, C57BL/6 mice are more susceptible than Balb/c mice to NAFLD/NASH development through genetic or epigenetic mechanisms influencing macrophage activation.71 Nevertheless, a comparison between seven WT mouse strains, which included a quantitative trait locus (QTL) analysis, identified the A/J strain as most susceptible to NAFLD development when fed with a MCD diet by exhibiting the highest serum alanine aminotransferase (ALT) levels and relatively high hepatic triglyceride content. In addition, for ALT and liver weight, QTLs on numerous chromosomes were identified, once more demonstrating the polygenic nature of NAFLD and NASH.72 QTL analyses are beneficial to identify chromosomal loci that control even small physiological effects in polygenic diseases such as NAFLD and NASH. When effects are measured in F2 intercrosses and compared with their parental inbred strains, QTL analyses are even more powerful and generate an overall picture of the number of QTLs that are segregating.73 Such QTL analyses on hepatic fibrosis in inbred mice and respective F2 intercrosses identified several loci that are involved in hepatic fibrosis, e.g. hepatic fibrogenic gene 1 and 2 and complement factor 5.74-76 An additional interesting approach combined a choline-deficient and L-amino acid defined diet with HF diet feeding and compared C57BL/6J (most prone to NAFLD development upon HF diet feeding, see the following) to A/J mice. Steatosis and elevated serum ALT levels were observed in both strains, while inflammation was minor in A/J mice and fibrosis could only be induced in C57BL/6J mice.47
When two mouse strains, C57BL/6 and 129Sv, were exposed to a HF diet, the mice exhibited similarities as well as substantial differences in metabolic and hepatic response. Both strains showed increased weight, serum cholesterol levels, and steatosis, while serum triglyceride levels were reduced. Differences between the two strains were obvious in the development of various metabolic features, for example C57BL/6 mice exhibited a more severe phenotype of obesity, glucose intolerance, and insulin response. Higher hepatic triglyceride accumulation and lower serum triglyceride levels were also observed in C57BL/6 mice compared with 129Sv mice.50,64,77 Moreover, 129Sv mice developed features of MS and steatosis only when fed the HF diet, while the C57BL/6 strain also showed a response to the low-fat (LF) control diet. The observed differences between the two strains may be due to an induction of SREBP-1c and stearoyl-coenzyme A desaturase-1 (SCD-1) expression and activity in C57BL/6 mice. These results implicate a role for dietary fat content and genetic predisposition in the development of NAFLD by SREBP-1c and SCD-1 action.77 A comparison of C3HeB/FeJ, C57BL/6NTac, C57BL/6J, and 129P2/OlaHsd fed with a HF diet gained dissimilar results; 129P2/OlaHsd and C3HeB/FeJ exhibited macrovesicular steatosis associated with a high hepatic triglyceride accumulation, and only microvesicular steatosis was exhibited in the two C57BL/6 strains. However, the conflicting results achieved by this study may be explained by differences in the composition of the used HF diet and treatment duration as well as genetic variations among the used mouse strains.78
A comparison of C57BL/6 and DBA/2 mice fed with a Western-type diet for 16 weeks also resulted in significant differences in susceptibility to NAFLD development. C57BL/6 mice exhibited a more severe degree of steatosis, including increased hepatic triglyceride levels and a greater peripheral insulin resistance compared with DBA/2J mice, though both strains developed obesity and severe hepatic insulin resistance. These observations suggest that peripheral rather than hepatic insulin resistance determines the degree of hepatic steatosis, and that development of peripheral insulin resistance may be determined by genetic factors that influence the susceptibility of different WT mouse strains to develop steatosis. Furthermore, development of obesity seems to be independent from genetic factors.79 Another study compared over 100 mouse strains for their susceptibility to HF and high-sucrose diet-induced NAFLD and found great differences in hepatic triglyceride accumulation. These differences may be caused by mitochondrial function, as well as the gut microbiome.80
Lastly, a study investigating 10 inbred mouse strains observed that even on a LF diet, hepatic triglyceride content varied due to the genetic background of the mice. Balb/c mice exhibited the highest hepatic triglyceride levels, most probably due to a short-chain acyl-CoA dehydrogenase deficiency, resulting in impaired β-oxidation of short-chain FAs. C57BL/6 mice showed an intermediate hepatic triglyceride accumulation and SWR mice displayed the lowest hepatic triglyceride content. Hepatic lipogenesis and triglyceride secretion were reduced in these mice while FA oxidation was higher than in both Balb/c and C57BL mice, suggesting that an increased hepatic triglyceride export protects SWR mice from hepatic triglyceride accumulation. Moreover, SWR mice appear to have low rates of lipolysis in adipose tissue, indicated by decreased plasma free FA levels when compared with Balb/c and C57BL mice, leading to decreased hepatic triglyceride production. This study also showed that C57BL mice expressed high levels of SCD-1, but nevertheless increased export and FA oxidation seem to restrain hepatic triglyceride contents, compared with Balb/c mice with lower SCD-1 expression. Although this study proves that genetic factors profoundly influence hepatic triglyceride accumulation, the factors and their contributing effects remain undetermined.81 Susceptibility of different commonly used WT mouse strains to the development of NAFLD/NASH features is summarised in Table 2.
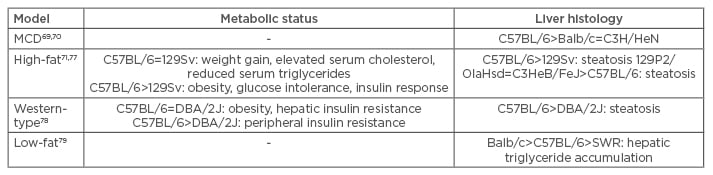
Table 2: Susceptibility of different common mouse wild-type strains to develop features of NAFLD/NASH.
MCD: methionine and choline-deficient.
In NAFLD/NASH research, the C57BL/6 strain represents a widely used WT model. For this strain, a heterogeneous metabolic response to HF diet feeding was reported.28,82,83 When C57BL/6 mice were fed a HF diet, the mice split in different metabolic subgroups, including lean non-diabetic, lean diabetic, and obese diabetic individuals.83 Also, in respect to hepatic injury, different phenotypes were observed.28,29,84 Thereby, the fat content of the used diets determined the development of liver disease in C57BL/6 mice. When fed a LF diet some C57BL/6 mice exhibited normal liver histology, while others developed benign hepatic lipid accumulation. HF diet feeding also resulted in two different hepatic phenotypes, including mice with macrovesicular steatosis and mice with more severe liver injury, exhibiting ballooning, Mallory–Denk body formation, and inflammatory cell infiltration in addition to steatosis.28 Furthermore, heterogeneous occurrence of fibrosis due to a HF, high-fructose, and high-cholesterol diet was reported.29
GENDER AND AGE EFFECTS ON NON-ALCOHOLIC FATTY LIVER DISEASE/NON-ALCOHOLIC STEATOHEPATITIS DEVELOPMENT AND PROGRESSION IN WILD-TYPE MICE
Another important aspect of NAFLD/NASH development is gender. For humans, it was shown that women have a higher prevalence of progression from NAFLD to NASH due to higher fibrotic activity.85-87 Such gender-specific differences in the development of NAFLD/NASH have also been reported in mice. A study in rodents fed a MCD diet compared not only different species and strains but also gender, and found an increased susceptibility to NAFLD/NASH among male rodents.48 Mice fed with diets rich in carbohydrates exhibited differences in the degree of inflammation; males showed steatosis in combination with inflammation, whereas females developed steatosis without signs of inflammation.88 Additionally, it was shown that dietary cholesterol content influences hepatic triglyceride accumulation, especially in female mice.30
Similarly, ageing may affect the development and progression of NAFLD/NASH in humans and mice.89-93 In humans, age only seems to be a risk factor for NAFLD in females.94,95 Nevertheless, age increases the risk of progression to steatohepatitis, fibrosis, and mortality.10,96-98 Similar observations were made for rodents. Studies comparing young, middle-aged, and old C57BL/6 mice fed a HF diet have found more liver damage and inflammation in the older mice, although the development of steatosis and the metabolic status were similar in all age groups.91,92 Another study on different-aged C57BL/6 mice showed that hepatic triglycerides significantly accumulate in older mice and that lipogenic genes are upregulated, even on a LF diet.93 However, the gender and age aspect remains controversial in both humans and mice, and therefore is a point of discussion in the published literature.
CONCLUSION
Dietary treatment in mice, with either methionine and choline restriction or over-nutrition, is a powerful model for human NAFLD and NASH, and will possibly help to refine diagnosis, prevention, and treatment of the diseases. Nevertheless, all dietary mouse models possess limitations, either in lacking the metabolic context of the human disease or in progression of NAFLD to NASH, as well as inconsistency between different approaches. The latter limitations may exist not only because of different formulation of the diets, but also because of different genetic backgrounds, varying age, and the respective gender of the mice used. Thus, more focus should be directed towards the genetic, age, and gender differences of the WT mice and their implication in the susceptibility to develop NAFLD/NASH, which may generate new insights in genetic determinants of human NAFLD and NASH.