Abstract
Chronic liver diseases take many forms; alcohol-related liver disease (ALD) and nonalcoholic fatty liver disease (NAFLD) are two common illnesses that potentially lead to cirrhosis, liver failure, and liver cancer. It is estimated that a quarter of heavy drinkers develop ALD and the same portion of people without heavy drinking habits have NAFLD. Alcohol intake is regularly used to differentiate NAFLD from ALD; however, diagnosis based on the discrimination threshold may be suboptimal when facing an obese patient with a high level of alcohol exposure. Therefore, understanding the common and/or different mechanism(s) driving each disease is extremely important. The ‘two-hit’ or ‘multi-hit’ hypothesis is used to explain the pathogenesis of both diseases. The ‘first hit’ refers to developing steatosis, the accumulation of fat components in the liver, and the ‘second hits’ are factors leading to oxidative stress, inflammation, and fibrosis, such as metabolic syndromes (e.g., morbid obesity, hyperglycaemia, hyperlipidaemia, disturbed circadian cycles, and altered intestinal microbiota) and environmental toxins (e.g., cigarette smoke and pollutants). Heritable factors also affect the probability and disease progression of both ALD and NAFLD. Whereas PNPLA3 and TM6SF2 variants are influential genetic risk factors for the diseases, epigenetic factors, such as DNA methylation, post-translational histone modifications, and small non-coding RNA, are of paramount importance. Moreover, considering that both ALD and NAFLD patients may eventually develop end-stage liver disease and require liver transplantation, the authors extensively investigated the worldwide outcomes from original literature for these two aetiologies, and the results showed no obvious differences in post-transplantation survival between them. Precise percentage determination of these two aetiologies contributing to steatohepatitis and its secondary injuries in the future would allow for better strategies for therapeutic and preventive intervention.
INTRODUCTION
Alcohol-related liver disease (ALD) and nonalcoholic fatty liver disease (NAFLD) are two very common types of chronic liver disease worldwide. ALD and NAFLD are multifactorial diseases with broad spectrums, including isolated steatosis (defined as hepatic triglyceride content [HTGC] >5%), steatohepatitis, fibrosis, and cirrhosis, and both can potentially lead to end-stage liver disease or develop into hepatocellular carcinoma (HCC),1 eventually requiring liver transplantation as a curative treatment. While ALD has a strong connection with heavy drinking, NAFLD can be found in >25% of adults that do not excessively consume alcohol in the USA.2 Generally speaking, NAFLD can be differentiated from ALD by using information about the alcohol consumption of the patient (<30 g per day for men and <20 g per day for women). However, the condition can be complicated when a patient has strong risk factors for NAFLD, such as Type 2 diabetes mellitus or obesity, while consuming excessive alcohol at the same time. Therefore, understanding the common and different mechanisms driving each disease is extremely important. In this literature review, the authors consider the different aetiologies in ALD and NAFLD to understand whether there are differences in post-transplant outcomes. The aim of this review is to compare the initial mechanisms, contributing factors, and liver transplantation outcomes between alcoholic steatohepatitis (ASH) and nonalcoholic steatohepatitis (NASH).
EPIDEMIOLOGIC FEATURES
Alcoholic Liver Disease and Nonalcoholic Liver Disease are Increasingly More Prevalent
Currently in the USA, chronic hepatitis C virus (HCV) is the top cause of liver decompensation leading to liver transplantation, followed by ALD. NASH is an advanced form of NAFLD and is more commonly associated with liver fibrosis compared with NAFLD outcomes. NASH is the third most common disease requiring liver transplantation.3 As one of the leading causes of hepatic decompensation, ALD is the second most common indication for liver transplantation and accounts for 40% of deaths from cirrhosis in Western countries.4 Studies show that 25% of heavy drinkers develop ASH, which is an advanced form of ALD, and, among them, about 40% will develop cirrhosis.5
On the other hand, the proportions of adults in the USA with nonalcoholic steatosis, NASH, and NASH-related cirrhosis or HCC are estimated to be 25%, 5–6%, and 1–2%, respectively.6,7 Such data suggest that in spite of the high prevalence of NAFLD, only a small portion of patients with nonalcoholic steatosis will eventually progress to end-stage liver disease. Nevertheless, considering the growing number of morbid obesity and metabolic syndromes, as well as effective treatment options for HCV eradication (direct-acting antiviral drugs), NASH will probably be the most common indication for liver transplantation in the near future.8 It is estimated that by 2025, 25 million Americans will have NASH, with 5 million of them developing cirrhosis or HCC.9
PATHOGENESIS
The ‘Two-Hit’ Hypothesis Can Apply to Both Alcoholic Steatohepatitis and Nonalcoholic Steatohepatitis
Isolated hepatic steatosis is 3–4-times as prevalent as NASH,10 and the key features differentiating NASH from isolated steatosis are the presence of cell injury and death.11 Much work has been done to identify definite mechanisms driving ALD and NAFLD disease spectrums. Obesity and extensive alcohol use are widely known risk factors related to fatty liver disease; however, only a minority of isolated steatosis cases will progress to steatohepatitis, cirrhosis, and HCC. Thus, the phenomenon is more likely a result of multifactor interaction. The ‘two-hit’ hypothesis, which was proposed by Day and James 20 years ago, is currently the leading hypothesis describing the pathogenesis of NASH as well as ASH.12 The ‘first hit’ refers to steatosis, the excessive accumulation of free fatty acids and triglycerides in hepatocytes. Earlier research has proven that many kinds of lipid-related intermediates are cytotoxic (lipotoxic) alone or together with the presence of other factors (‘second hits’) and cause damage to hepatocytes through lysosomal dysregulation and elevation of endoplasmic reticulum stress.13,14 An example of lipotoxicity demonstrated both in vivo and in vitro was that deactivation of stearoyl-CoA desaturase-1, the enzyme that converts saturated fatty acids to monounsaturated fatty acids, sensitised hepatocytes to monounsaturated fatty acid-induced caspase activation and apoptosis (Table 1).15
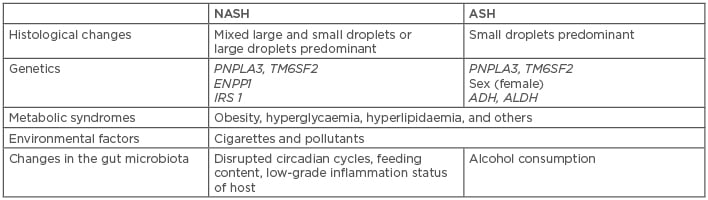
Table 1: Brief comparison of pathogenic causes of nonalcoholic steatohepatitis and alcoholic steatohepatitis.
ADH: alcohol dehydrogenase; ALDH: aldehyde dehydrogenase; ASH: alcoholic steatohepatitis; ENPP1: ectonucleotide pyrophosphate/phosphodiesterase 1; IRS 1: insulin receptor substrate 1; NASH: nonalcoholic steatohepatitis; PNPLA3: patatin-like phospholipase domain-containing 3; TM6SF2: transmembrane 6 superfamily, member 2.
The Mechanisms of Steatosis are Closely Associated with Insulin Resistance
The livers of patients with NAFLD usually contain mixed large (macrovesicular) and small (microvesicular) droplets or predominantly large droplets within hepatocytes, while the livers of patients with ALD are more predominantly microvesicular (known as alcoholic foamy degeneration).16 Such pathologies may be due to different fat accumulation processes in ALD and NAFLD and could be helpful in determining the percentage of contribution of ALD and NAFLD. In the case of NAFLD, insulin resistance and consequent hyperinsulinaemia, which causes lipolysis of peripheral fat and increased fat absorption of the liver, are thought to be the triggers of steatosis. However, in the case of ALD, the definite mechanisms of steatosis are more complicated, given the fact that ethanol has dose-dependent effects on insulin signalling: high-dose ethanol exposure weakens insulin signalling in hepatocytes through elevation of tribbles- related protein 3, leading to decreased sterol regulatory element binding protein-1. In contrast, low-dose ethanol consumption enhances hepatic insulin signalling associated with reduced p55γ (a phosphatidylinositol 3-kinase regulatory subunit isoform) to increase nuclear sterol regulatory element binding protein-1.17 Even so, the protective effect of low-dose alcohol exposure is controversial. Bellentani et al.18 claimed that patients with NAFLD should not seek a preventive effect via drinking a small amount of alcohol on a daily basis because “the alcohol and the metabolic risk factors for progression of liver disease should be considered always together”, implying that, in many cases, the steatohepatitis may not be purely alcoholic or nonalcoholic but rather a mixed coexistance of both ALD and NAFLD.
Metabolic Syndromes and Environmental Toxins Act as Second Hits Toward Steatohepatitis
The second hits, including obesity, hyperglycaemia, hyperlipidaemia, and other metabolic syndromes, as well as environmental toxins, such as cigarette smoke and pollutants, lead to oxidative stress, inflammation, and fibrosis, which are important drivers of the pathogenesis and progression of steatohepatitis.19,20 Oxidative stress could increase the generation of cytokines such as TNF-α, transforming growth factor beta, and Fas ligand, participating in the progression of steatohepatitis, such as cell death, further inflammation, and fibrosis.21-23 To protect themselves from further damage, injured hepatocytes undergo adaptations known as stress responses, which rebalance signalling cascades and other aspects of cell physiology. As a result of the unstable microenvironment, the hepatocytes are more vulnerable to other stimulants and stressors, which may also cause apoptosis. For example, in mice models, >50% loss of glucose-regulated protein 78, which is a master regulator of endoplasmic reticulum homeostasis, causes compartment dilation in endoplasmic reticulum, as well as other stress-induced responses including fat accumulation, insulin resistance, increased susceptibility to alcohol, overfeeding, and drug and toxin-related injury, leading to cell apoptosis.24 Dying cells then produce mediators that promote the regeneration process to replace themselves. In a liver undergoing active dying and regenerating processes, regenerative cell types, including immune cells, myofibroblasts, and hepatic progenitor cells, will be present in a larger amount than those in a normal, healthy liver.25,26 The first and second hits can interact with each other, causing hepatocyte injury, and the dying and regenerating processes finally become futile, resulting in progressive scarring, which leads to steatohepatitis, fibrosis, cirrhosis, and liver cancer, resulting in hepatic decompensation.
Circadian Cycles and Intestinal Microbiota are Involved in Steatohepatitis Pathogenesis via the Gut–Liver Axis
Until recently, the link between stressful daily routines and liver diseases was unclear, but new experimental and clinical evidence has revealed the interaction between circadian cycles, gut microbiota, intestinal barrier integrity, and NAFLD.27 Normally, intestinal microbes and the host share a mutually symbiotic relationship, forming a complex and diverse ecosystem. When the host experiences disrupted circadian cycles, which are often seen in patients with shift work and frequent long distance travel, the normal intestinal flora will be influenced rapidly.27 Feeding content and frequency, as well as chronic, low-grade inflammation status of the host, accompanied by altered inflammatory cytokines, such as elevated TNF-α and IL-6, impact intestinal microbiota as well.28 Altered intestinal microbiota lose the normal function of intestinal wall barriers, leading to increased intestinal permeability and further production of fatty acids in the small bowel, causing additional fatty acid absorption, thereby strengthening the first hit and resulting in obesity and obesity-related steatohepatitis.29 Besides this, altered intestinal microbiota dysregulate hepatic inflammation via supplement of bacterial endotoxins and other toll-like receptor ligands, which induce hepatocytes to produce inflammatory mediators, including TNF-α, IL-1, and plasminogen activator inhibitor-1.23 In short, host factors (diet content, feeding frequency, biological clocks, and chronic inflammation) influence normal gut flora, and, conversely, abnormal gut flora affect the host, leading to pathological inflammation and NASH. Such cross-talk between the liver and the gut, known as the gut–liver axis, are bidirectional actions that play a critical role in NASH pathogenesis.
In the case of ASH, recent animal studies by Lowe et al.30 showed a link between alcohol, gut microbiota, hepatic neutrophil infiltration, and hepatitis. In their study, changes in gut microbiota were found in alcohol-fed mice and were related to hepatic neutrophil infiltration, inflammation signalling, and steatotic changes. Elevated serum alanine and aspartate aminotransferase, and activation of fat metabolic signalling pathways, were also discovered. Suppression of gut bacterial load by antibiotics reduced alcohol-related liver steatosis but not serum transaminases. In human cohorts, Dubinkina et al.31 demonstrated an association between alcohol consumption, gut microbiota, and liver dysfunction. Increased gut permeability, impaired ability to transform toxic ethanol metabolites, such as acetaldehyde, and dysregulation of bile volume and composition are all possible mechanisms linking alcohol consumption and liver disease.
Role of Genetics: Vulnerable Background for ‘Hits’
Heritable components play roles in determining the risks of both ALD and NAFLD. Sex is a well-known risk for developing ALD: among people who have similar levels of ethanol exposure, women are more susceptible to ALD than men.32 Enomoto et al.33 demonstrated in a rat model that oestrogen promotes the production of TNF-α via increasing the permeability of the intestinal wall to endotoxins and upregulating endotoxin receptors on Kupffer cells. Moreover, twin studies have shown that ALD was three-times higher in monozygotic twins than in dizygotic counterparts.34 On the other hand, familial aggregation studies indicated that genetic factors play roles in developing NAFLD.35,36 Much work had been done in recognising the associated genetic and epigenetic factors regarding the risk of NAFLD.
PNPLA3, TM6SF2, and Other Genetic Variants are Risk Factors for Alcoholic Liver Disease and Nonalcoholic Fatty Liver Disease
Many gene polymorphisms have been identified as strongly relevant to either ALD or NAFLD, including PNPLA3 variants and TM6SF2 variants, that promote the development of steatohepatitis and further hepatic injury.5,37 A statistically significant index single-nucleotide polymorphism (SNP) in PNPLA3 (rs738409, I148M), which is more prevalent in Asian and Native American populations, was identified as being associated with higher hepatic susceptibility to both alcoholic and nonalcoholic steatosis, steatohepatitis, cirrhosis, and liver cancer.38,39 Combined with another SNP (rs6006460, p.S453I) in the same gene, these variants lead to even further increases in HTGC, increasing the severity of ALD and NAFLD disease spectrums.39 TM6SF2, located within the 19p13.11 locus, is responsible for encoding a protein that regulates liver fat metabolism, influencing triglyceride secretion and hepatic lipid droplet content. A TM6SF2 variant (rs58542926, p.E167K), which downregulates hepatocyte very-low-density lipoprotein secretion, is associated with a higher risk of increased HTGC, leading to steatohepatitis, fibrosis, and further liver damage.40 However, neither PNPLA3 nor TM6SF2 variants are necessary for development and progression of steatohepatitis.19,37 In addition, other aetiology-specific genetic polymorphisms are only related to either ALD or NAFLD independently. For example, variants in genes encoding class I alcohol dehydrogenase and aldehyde dehydrogenase alter the activity of the enzymes, thus potentially determining ethanol tolerant ability, risk of addiction to alcohol, and risk of ALD,41,42 while variants in genes encoding ectonucleotide pyrophosphate/phosphodiesterase 1 and insulin receptor substrate 1 impair insulin signalling, thus elevating the risk of nonalcoholic steatosis and NASH.43
Epigenetic Factors Affect the Probability of Developing Steatohepatitis, Directly or Indirectly
DNA methylation, post-translational histone modifications, and RNA-based mechanisms (e.g., small non-coding RNA [miRNA]) are important epigenetic factors that regulate gene expression without altering the primary DNA sequences. Some studies suggested that inactivation of some tumour-suppressor genes, due to promoter DNA hypermethylation, is responsible for liver fibrosis and cancer. For example, phosphatase and tensin homologue, a tumour suppressor gene frequently inactivated on chromosome 10q23, inhibits extracellular signal regulated kinase and protein kinase B pathway and downregulates cell cycles and proliferation.44,45 miRNA, which are small, non-coding and single-strand RNA comprising 19–22 nucleotides, are involved in the regulation of cell development, proliferation, differentiation, and apoptosis through a variety of translation regulatory functions. Silencing of certain miRNA by promoter hypermethylation is related to the regulatory pathways involved in inducing liver fibrosis (e.g., MeCP2, a member of methylated DNA-binding domain proteins, and DNA methyltransferases family-related pathways).44,46 Moreover, correlation between the risk of obesity and metabolic syndromes in children and mothers experiencing starvation was noted through epigenetic modification, potentially leading to the formation of NAFLD.47 Such epigenetic factors as well as genetic components directly or indirectly influence the probability of steatohepatitis and further liver damage.
OUTCOMES OF LIVER TRANSPLANTATION AS THE ULTIMATE THERAPEUTIC TREATMENT: CURRENT STATUS AND A LOOK TOWARDS THE FUTURE
Liver transplantation is the ultimate treatment choice for decompensated livers. As mentioned above, ASH and NASH are the second and third most common indications for liver transplantation, respectively, following HCV.3,8 Nowadays, effective treatment for liver chronic viral infection has been well developed; therefore, in the near future, ASH and NASH are set to become the two most common indications for liver transplantation. With this expectation in mind, there are two important questions the medical community needs to address: are the transplantation outcomes of ASH and NASH patients similar? Secondly, are the disease recurrence rates between ASH and NASH similar?
Bhagat et al.48 compared the 1-year, 3-year, 5-year, and 9-year post-transplant survival between ASH and NASH patients and reported no significant difference (p=0.17). In their study, sepsis with or without multiorgan failure was the leading cause of mortality in both ASH and NASH groups, followed by malignancies in the ASH group and cardiovascular causes, including myocardial infarction and stroke, in the NASH group.48 In order to have a broader view, the authors of this paper extensively reviewed original literature regarding liver transplantation outcomes of either ASH or NASH as the indication (Table 2 and Table 3)48-63 from 1982–2010. Studies that did not distinguish ASH and NASH from other liver diseases, such as HCV or autoimmune hepatitis, were excluded.
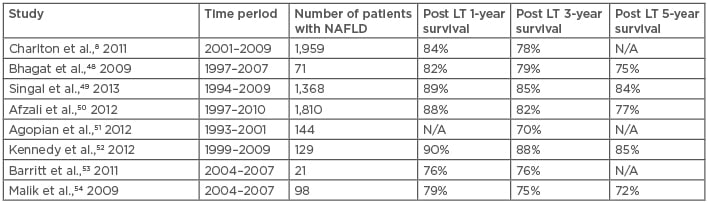
Table 2: Outcome of liver transplantation for nonalcoholic steatohepatitis.
LT: liver transplant; N/A: not applicable; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis.
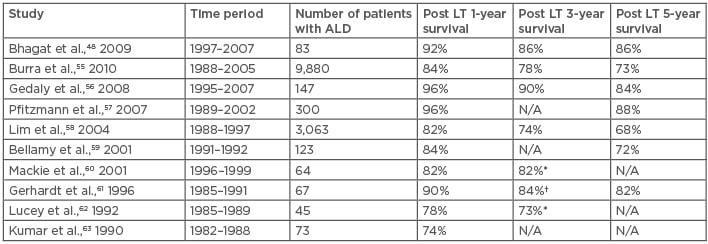
Table 3: Outcome of liver transplantation for alcoholic steatohepatitis.
*2–year survival; †2–4-year survival
ASH: alcoholic steatohepatitis; ALD: alcoholic liver disease; LT: liver transplantation; N/A: not applicable.
Post-Transplantation Survival Between Alcoholic Steatohepatitis and Nonalcoholic Steatohepatitis Patients are Similar Worldwide
Although the comparison may not be totally valid because the patients were observed and followed-up in different centres and during different eras, the results of this review still provide a complete picture, as different studies showed similar trends. Survival at 1, 3, and 5 years after liver transplantation for NASH was 76–90%, 75–88%, and 72–85%,8,48-54 compared with 74–92%, 73–90%, and 72–88% for ALD,48,55-63 respectively. It could be summarised that outcomes for patients receiving liver transplantation for ASH and NASH are similar. However, Bhagat et al.48 reported a significantly higher post-transplant recurrence rate of steatohepatitis in livers with NASH than with ASH (33% versus 0%; p<0.0001) but no significant difference in retransplantation rates. Such outcomes may be a result of the different initial mechanisms of steatohepatitis. In addition, active alcohol abuse is an absolute contraindication for liver transplantation whereas metabolic syndrome is not; in other words, the underlying conditions leading to NAFLD are not eliminated when a patient with NASH receives liver transplantation. However, given the fact that alcohol consumption is contraindicated, many ASH patients experience ‘alcohol relapse’, with a relapse rate of 10–50%.64 Further study is needed to clarify factors that determine the prognosis. Both ASH and NASH patients could benefit from liver transplantation with no significant survival difference and, considering underlying ‘abnormal metabolic’ status, there may be a role for aggressive control of metabolic syndrome in post-transplant NASH patients. As for ASH patients, cardiovascular disease and de novo malignancies seem to have great impacts on patients’ quality of life and survivability, and thus they should be taken into consideration by the transplant team.64
CONCLUSION
ALD and NAFLD are increasingly important chronic liver diseases that lead to advanced liver damage, such as cirrhosis and liver cancer. While the alcohol volume threshold is widely used to distinguish NAFLD from ALD, there may be a combination of causes in many cases, considering that many people eat and drink a lot at the same time. ALD and NAFLD share many common pathological pathways as well as similar presentation. The two-hit hypothesis could apply to the pathogenesis of both ALD and NAFLD, with the first hit being steatosis, the excessive accumulation of fat components in hepatocytes, and the second hits being the stressors that cause the progression of steatohepatitis. Altered intestinal microbiota is a newly recognised risk factor leading to NASH and ASH via the cross-talk between the liver and the gut. Additionally, a number of genetic polymorphisms, including PNPLA3 and TM6SF2 variants, as well as epigenetic factors such as DNA methylation, post-translational histone modifications, and RNA-based mechanisms, increase the risk of ALD and NAFLD and affect the severity and progression. Although the complexity of steatohepatitis aetiologies results in great challenges in prevention and treatment, new drugs targeting inhibition of the inflammatory process and the improvement of insulin signalling are being tested in patients with NASH.65 With the increasing number of patients requiring liver transplantation with ASH and NASH as aetiologies, there seems to be no significant difference in outcomes for these patients, and such results are consistent with previously published data.48
Currently, there are no clinical examination techniques that are able to definitely distinguish NASH from ASH or determine the precise percentage of contribution in each patient. The authors believe that since steatohepatitis is a multifactorial condition, people with steatohepatitis probably have traits of both ASH and NASH. In other words, both diseases may coexist within one liver but consist of different spectrums. A precise diagnostic tool could allow physicians to formulate better therapeutic or preventive strategies. For example, for an ASH-dominant liver, quitting drinking is the best solution, whereas for a NASH-dominant liver, controlling body weight and blood sugar may be more efficient. Diagnosis based on drinking volume alone is not sufficient. Combining a specific set of biomarkers, genetic information, dynamic liver testing, and advanced imaging techniques may be promising new solutions for the diagnosis of steatohepatitis in the future.66