Meeting Summary
This symposium was held on the first day of the 2023 European Hematology Association (EHA) Congress, held in Frankfurt, Germany. The main objective of the symposium was to gather experts from the field to raise awareness of the challenges in diagnosing and treating systemic mastocytosis (SM) with an associated haematologic neoplasm (AHN). Presentations focused on optimising the diagnosis of the SM component and recognising the different types of myeloid AHN. The expert panel considered the clinical management of SM-AHN, and how and when to prioritise the various disease components.
The overarching message from the symposium was that diagnosis of SM-AHN is challenging, and SM is often missed in patients with a myeloid neoplasm, such as chronic myelomonocytic leukaemia (CMML), myelodysplastic/myeloproliferative neoplasm (MDS/MPN), myelodysplastic syndrome (MDS), and myeloproliferative neoplasm (MPN), as well as AHN being missed in patients with SM. Identification of a SM-AHN through serum tryptase and/or molecular testing for KITD816V mutation in peripheral blood in a patient with a previous diagnosis of a myeloid neoplasm allows potentially efficacious targeted treatment with KIT inhibitors, such as midostaurin and avapritinib. Although SM-AHN is associated with a poor prognosis, a correct diagnosis and detailed understanding of an individual’s disease can help to guide optimal treatment decisions, including when to prioritise SM treatment over AHN treatment, and vice versa.
Introduction
Julien Rossignol, Andreas Reiter, and Deepti Radia
Julien Rossignol, Haematologist at Centre de référence de mastocytoses (CEREMAST), a national reference centre for mastocytosis at the Hôpital Necker Enfants Malade, Paris, France, described mastocytosis as a group of rare myeloid neoplasms characterised by the accumulation of atypical, KITD816V-mutated (mostly clonal) mast cells, including cutaneous mastocytosis, SM, and mast cell sarcoma. Up to 10% of all cases of SM can be classified as advanced SM,1,2 a category that includes SM-AHN, aggressive systemic mastocytosis, and mast cell leukaemia.3-5 The prognosis for advanced SM is poor, with a median survival of 24–42 months,6 mainly due to the abnormal infiltration of mast cells into various organs resulting in severe and debilitating symptoms, and life-threatening organ damage.5,7
Approximately two in three patients with advanced SM also have an AHN of myeloid origin.7 Andreas Reiter, Professor at the Centre of Excellence for Mastocytosis, University Hospital Mannheim, Heidelberg University, Germany, mentioned that approximately 5–10% of patients with CMML, approximately 2–5% patients with MDS/MPN, and approximately 2–3% of patients with MPN or acute myeloid leukaemia (AML) may carry the KITD816V mutation (Reiter, personal communication based on the German Registry on Disorders of Eosinophils and Mast Cells, unpublished data). Moreover, it is one of the most frequent molecular abnormalities in the diagnostic work-up of eosinophilia.8 Based on a retrospective study, the majority of patients (91%) with KITD816V-mutated chronic myeloid neoplasms, such as CMML, MDS/MPN, MDS, or MPN, had concurrent SM, which has been shown to be missed in nearly one-third of cases.9
Deepti Radia, Consultant Haematologist at Guy’s and St Thomas’ Hospitals NHS Foundation Trust, London, UK, explained that SM-AHN has a complex, heterogenous clinical presentation, involving the SM variant, SM burden, AHN subtype, and AHN risk profile.3,10,11 This can make accurate diagnosis of advanced SM challenging, yet it is extremely important because of the substantial impact it has on patient management decisions.2
Which Myeloid Neoplasms Occur in Patients with Systemic Mastocytosis with an Associated Haematologic Neoplasm?
Julien Rossignol
The vast majority of SM-AHN are associated with myeloid neoplasms rather than lymphoid neoplasms, such as lymphoma or multiple myeloma.3,5 Rossignol explained that this may be because of the clonal architecture of SM-AHN associated with additional mutations in myeloid neoplasms, particularly MDS, MPN, and MDS/MPN. This is suggested by several studies, which found that mutations associated with AHN affect genes such as TET2, SRSF2, and ASXL1, and that KITD816V, which is frequently associated with SM, is a sub-clonal mutation, occurring later in the hierarchy of differentiation.5,9,12 Rossignol mentioned that in the French national registry, CMML is the most frequent AHN associated with SM, followed by other MDS/MPN, MPN, and MDS. The SM component is frequently advanced, especially in patients with MDS/MPN (Rossignol, personal communication based on the French national registry, unpublished data).
Optimising Diagnosis and Subtyping in Systemic Mastocytosis with an Associated Haematologic Neoplasm
Andreas Reiter, Eric Solary, and Julien Rossignol
Reiter highlighted that approximately 20% of cases of SM may initially be missed in patients with chronic myeloid neoplasms, leading to delayed diagnosis and treatment for patients.13 Reiter explained that SM should be suspected in patients presenting with anaphylaxis, flushes, fatigue, typical skin lesions, or symptoms in bones or the gastrointestinal tract. Reiter highlighted that routine tryptase testing in blood (or tryptase staining in bone marrow biopsy) and high-sensitivity KITD816V variant allele fraction testing in peripheral blood can help to screen for hidden SM in myeloid neoplasms. Eric Solary, Professor of Haematology at the Gustave Roussy Cancer Center and Paris-Saclay University, Villejuif, France, recommended that AHN should be suspected in patients diagnosed with SM when there is evidence of monocytosis, eosinophilia, splenomegaly, elevated lactate dehydrogenase, high KITV617F variant allele fraction in peripheral blood, or additional somatic mutations.
The 2022 World Health Organization (WHO) and International Consensus Classification (ICC) criteria for the diagnosis of SM are very similar.4,14 For SM diagnosis, a patient needs to meet one major and one minor, or three minor criteria. The major criterion for diagnosis of SM is the presence of “multifocal dense infiltrates of mast cells (≥15 mast cells in aggregates) in bone marrow biopsies and/or in sections of other extracutaneous organ(s) component of SM.”4,14 However, Rossignol stressed that bone marrow biopsy is not routinely performed in all countries, and where it is performed, it may be challenging for an inexperienced pathologist to recognise mast cells in bone marrow biopsy if not using appropriate mast cell biomarkers, such as tryptase and cluster of differentiation 117 (CD117). Rossignol suggested that these issues may contribute to the underdiagnosis of the SM component of SM-AHN.
One of the four minor criteria for the diagnosis of SM is the detection of a “KIT-activating KIT point mutation in codon 816 or in other critical regions of KIT in bone marrow or another extracutaneous organ.”4,14 Rossignol explained that next-generation sequencing has a low sensitivity for detecting KIT mutations, because of the low KITD816 variant allele fraction in the peripheral blood of many patients, and they recommended the use of a high-sensitivity assay, such as droplet digital PCR, for this purpose.15,16
Another minor diagnostic criterion for SM is the detection of “baseline serum tryptase concentration >20 ng/mL.”4,14 Rossignol pointed out that tryptase is not routinely assessed in patients diagnosed with myeloid neoplasms (especially those with CMML), and suggested that this test should be more commonly performed to screen for SM in this population, in order to alert the clinician to the possibility of concurrent SM disease.
SM-AHN should be further classified according to AHN type.3,4,14 The AHNs most frequently associated with SM are MDS/MPNs, particularly CMML.9
The 2022 WHO and ICC definitions of CMML are similar, incorporating blood monocytes at ≥500 cells/µL and ≥10% of white blood cells; clonality markers; and (‘or’ in the ICC definition) cell dysplasia. In addition, the ICC definition requires an abnormal immunophenotype consistent with CMML.4,14 CMML-1 presents with <5% blast cells in peripheral blood and <10% in bone marrow, and CMML-2 with 5–19% blasts in peripheral blood and 10–19% in bone marrow.4
Solary described the typical CMML patient as a male of approximately 70 years of age, in whom monocytosis is revealed through a routine blood test. If subsequent tests reveal cell dysplasia, clonality, or an abnormal partition of monocyte subsets, then that patient should be diagnosed with CMML.4 Overall, CMML is a severe disease with median survival of approximately 29 months.17
Clinical Updates in Treatment of Systemic Mastocytosis with an Associated Haematologic Neoplasm
Andreas Reiter and Eric Solary
Systemic Mastocytosis Component
Prior to the development of novel tyrosine kinase inhibitors, targeting KIT D816V, such as multikinase inhibitor midostaurin and selective inhibitor avapritinib, treatment for patients with advanced SM was based on the off-label use of the purine analogue, cladribine.18 Despite remaining an off-label therapy, Reiter explained that cladribine is still a relevant treatment option for some patients.18
An analysis of registry data found that cladribine treatment in patients with advanced SM was associated with a median overall survival of 1.9 years at first-line (n=48) and 1.2 years at second-line (n=31), and risk stratification made little difference to these outcomes.18
In contrast, an open-label clinical trial found that midostaurin treatment of patients (N=116) with advanced SM was associated with a median overall survival of 2.4 years (28.7 months) and a median progression-free survival of 1.2 years (14.1 months). Moreover, for the first time, reductions in all measures of mast cell burden were observed, including bone marrow mast-cell burden, serum tryptase levels, and spleen volume. Midostaurin improved most symptoms except for nausea and vomiting, which were the most common non-haematologic adverse events (AE; experienced by 79% and 66% of patients, respectively).19 Reiter noted that most patients achieved partial responses on midostaurin. Based on these data, midostaurin was approved in 2017 as a first tyrosine kinase inhibitor for the treatment of patients with advanced SM.20
In the absence of randomised trials in this rare disease setting, a retrospective comparative analysis of data from the German Registry on Disorders of Eosinophils and Mast Cells (GREM) for patients with advanced SM treated with midostaurin (n=63) and cladribine (n=23) showed that midostaurin was associated with a significantly improved median overall survival versus cladribine (4.2 years versus 1.9 years, respectively; p=0.033), and a significantly higher probability of leukaemia-free survival (2.7 years versus 1.3 years, respectively; p=0.044) on the basis of a propensity score-weighted analysis.21
In 2022, a second tyrosine kinase inhibitor, avapritinib, was approved for treatment of patients with advanced SM after at least one systemic therapy.22 The safety and efficacy of avapritinib in patients with advanced SM was assessed in two open-label studies: EXPLORER7 and PATHFINDER.23 A post hoc analysis was conducted on the pooled sub-population of patients from these two studies who were initiated on 200 mg daily of avapritinib, and who had received ≥1 prior therapy (N=31).24 With a median duration of follow-up of 17.7 months, the overall response rate among patients with advanced SM was 71%, and in patients with SM-AHN, the most challenging subtype, the overall response rate was 77% (n=22). Reiter noted that for the first time, complete remission (3%) and complete remission with partial haematologic recovery (16%) was observed in patients with advanced SM. Mutations in SRSF2, ASXL1, and/or RUNX1 are associated with poor prognosis in advanced SM, but they had no apparent effect on avapritinib efficacy, with an overall response rate of 64% in patients with ≥1 mutation in SRSF2, ASXL1, and/or RUNX1; and 78% in patients without. Avapritinib also reduced all measures of mast cell burden: 89% of patients had ≥50% reduction from baseline in bone-marrow mast cell infiltrates (60% had total clearance of mast cell aggregates); 89% had ≥50% reduction from baseline in serum tryptase; 66% had ≥50% reduction from baseline in KITD816V variant allele fraction (21% to below the limit of detection); and 70% had ≥35% reduction from baseline in spleen volume. In terms of the AHN components of disease, avapritinib appeared to reduce eosinophil counts in all patients with baseline eosinophilia; and to normalise peripheral monocytes in almost all patients with baseline monocytosis.24
The most common AEs shown to occur with avapritinib 200 mg in advanced SM were periorbital oedema (38%), thrombocytopenia (37%), peripheral oedema (33%), and anaemia (22%).25 Among 193 patients enrolled in avapritinib studies for advanced SM (all doses), 12% experienced serious AEs during treatment.25 Among patients on 200 mg avapritinib (n=126), only 7% experienced an AE leading to permanent discontinuation, and 79% reduced the dose of avapritinib after a median of 6 weeks of treatment. Serious AEs of intracranial haemorrhage have been reported in patients with advanced SM receiving avapritinib, and therefore avapritinib is not recommended in patients with platelet counts <50×109 /L. Platelet counts should be regularly monitored during treatment, and low platelets can be managed by temporarily interrupting avapritinib and modifying the dose. Platelet support, including thrombopoietin agonists, may be considered in some cases (see avapritinib summary of product characteristics for further details).25
There are no randomised trials that compared avapritinib with alternative therapies in advanced SM, because the disease is so rare. Therefore, Reiter et al.26 compared clinical trial data for avapritinib (n=176) with real-world data from a multicentre, observational, retrospective chart review of patients who received systemic treatment for advanced SM (best available therapy cohort [including midostaurin and cladribine]; n=141). The study found that among patients with ≥1 prior line of therapy, those treated with avapritinib 200 mg had a 63% lower risk of death compared with those treated with best available therapy (p=0.006).26
Associated Haematologic Neoplasm Component
Given the clinical and biological heterogeneity of CMML, Solary explained that the current treatment approach ranges from ‘watch and wait’ with active monitoring, through erythropoiesis-stimulating drugs to treat anaemia, to more aggressive therapy in patients with proliferative disease and poor risk factors, including the use of cytoreductive drugs (e.g., hydroxyurea), hypomethylating agents (HMA; e.g., azacitidine and decitabine), or allogeneic haematopoietic cell transplantation (alloHCT), depending on the patients’ age and comorbidities.27
Regarding alloHCT, Solary shared that a large retrospective cohort study found that performing alloHCT before a patient progresses from CMML to acute AML can decrease the life expectancy in lower-risk patients (N=1,114; hazard ratio [HR]: 3.19; p<0.001), but it can be beneficial and may be considered in high-risk patients with CMML.28 Solary also presented data from two studies illustrating that HMA therapy does not impact the genetic component of disease in CMML. The first study showed that although an HMA can generate a complete clinical response, with correction of cytopenia, decrease in spleen volume, and reduction of monocytosis, it does not decrease the size of the mutated clone or prevent somatic evolution (N=17), and this can eventually lead to relapse.29 The second study showed that while treatment with decitabine significantly reduced the risk of progression to AML compared with hydroxyurea treatment (HR: 0.62), it increased the risk of death without progression/transformation (HR: 1.55; N=170). Therefore, HMA treatment did not improve event-free survival when compared to hydroxyurea in this population (HR: 0.83).30
Solary described CMML as a disease of ageing with a proliferative component, few residual wild-type stem cells, and alteration of the bone marrow niche. Solary stressed that the current treatment approaches do little to reduce the diseased cell count, and that dedicated treatments need to be developed for specific subsets of CMML. Solary posited that it might be prudent to approach treatment with an aim to slow disease progression and improve quality of life, rather than eradicating the clonal cells.
In general, there is a strong need for new therapeutic strategies in CMML, including optimising the use of epigenetic regulators (epidrugs), such as HMAs and combinations; developing new treatments for cytopenia (e.g., ligand traps for activins and growth differentiation factor 11, and eltrombopag for thrombocytopenia); and targeting granulocyte macrophage colony stimulating factor signalling, rat sarcoma virus (RAS) signalling, or other pathways and targets, such as the cytokines released by mature CMML monocytes and the cells of the niche.31
Challenges in Treatment Decision-Making for Systemic Mastocytosis with an Associated Haematologic Neoplasm
Deepti Radia
The complex, heterogenous clinical presentation of SM-AHM can make accurate diagnosis challenging (Figure 1). Radia explained that there are numerous variables that inform decisions on treatment strategy, such as the SM component subtype and symptom burden; the AHN component subtype, including mutations and lineage; the presence of organ involvement, comorbidity and age; and AHN risk category.3,4,32

Figure 1: Clinical presentation of systemic mastocytosis with associated haematologic neoplasm varies, and involves both systemic mastocytosis variant and burden, and associated haematologic neoplasm subtype and risk profile.3,10,11,13
Reproduced with permission from Blueprint Medicines.3,10,11,13
AHN: associated haematologic neoplasm; AML: acute myeloid leukaemia; ASM: aggressive systemic mastocytosis; BMM: bone marrow mastocytosis; CEL-NOS: chronic eosinophilic leukaemia, not otherwise specified; CMML: chronic myelomonocytic leukaemia; HES: hyper-eosinophilic syndrome; ISM: indolent systemic mastocytosis; MCL: mast cell leukaemia; MDS: myelodysplastic syndrome; MPN: myeloproliferative neoplasm; MPN-U: myeloproliferative neoplasm-unclassified; SM: systemic mastocytosis; SSM: smouldering systemic mastocytosis.
Radia shared their approach to guiding treatment pathways in SM-AHN, explaining that they are more likely to prioritise treatment of the SM component if it is associated with a high symptom burden, C-finding attributed to SM, high tryptase level, and high KIT mutation burden; and more likely to prioritise treatment of the AHN component if it is associated with monocytosis or eosinophilia, low KIT mutation burden, and altered bone marrow morphology with significant additional high-risk mutations ([hlFigure 2[/hl]).32
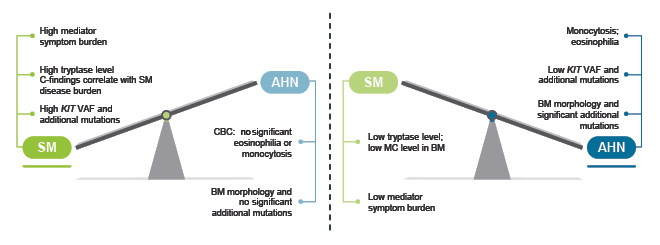
Figure 2: An approach to guiding treatment priorities in systemic mastocytosis with associated haematologic neoplasm.32
Reproduced with permission from Radia.32
AHN: associated haematologic neoplasm; BM: bone marrow; CBC: complete blood count; MC: mast cell; SM: systemic mastocytosis; VAF: variable allele frequency.
The remaining challenges and questions that need to be addressed for optimal treatment of SM-AHN were summarised by Radia as: what are the best approaches to risk stratification for individual patients; should we routinely use mutational profiles to guide treatment and monitor responses in all patients with SM; how and when should combination or sequential treatment be offered; and what is the place of alloHCT as a curative option?
Key Summary Points
• Myeloid neoplasms, such as CMML, MDS/MPN, MPN, and MDS, are most often found in association with SM (SM-AHN).
• The diagnosis of SM-AHN is frequently delayed: SM is missed in patients with myeloid neoplasm, or AHN is missed in patients with SM.
• Routine testing for serum tryptase and quantitative KITD816V analysis in peripheral blood using a high-sensitivity assay can help to screen for SM in patients with myeloid neoplasms, especially CMML.
• Identification of a SM-AHN in a patient with a previous diagnosis of a myeloid neoplasm allows potentially efficacious targeted treatment with midostaurin and avapritinib.
• Treatment decisions in SM-AHN depend on patient symptoms, mast cell burden, presence of C-findings, KITD816V lineage involvement, AHN burden, and additional somatic mutations.
• There is a risk of progression of AHN, e.g., secondary AML, with the need to monitor the clonal evolution during treatment.
• Sequential targeted therapy can be beneficial in the management of SM-AHN.