![EMJ Hematology 8 [Supplement 5] 2020 Feature Image](https://www.emjreviews.com/wp-content/uploads/2020/09/EMJ-Hematology-8-Supplement-5-2020-Feature-Image-2021-940x563.jpg)
Meeting Summary
The 25th European Hematology Association (EHA25) Annual Congress was conducted as a virtual meeting because of the coronavirus disease 2019 (COVID-19) pandemic. This article reviews several sessions from the EHA25 Congress covering updates in haematology. The first, ‘Sickle cell disease (SCD): update on emerging chronic complications’, reviewed the effect of cardiac complications, fertility with hydroxyurea treatment, and delayed haemolytic transfusion reaction in SCD. The second session focussed on topics relating to treatment for SCD, including iron overload and its management with chelation therapy, novel treatment options, and different treatments available worldwide, with a focus on the use of hydroxyurea in Africa. The third session, ‘Multiple perspectives on the burden and management of sickle cell disease’, including patient voices from the Sickle Cell World Assessment Survey (SWAY), provided an overview of effective management of vaso-occlusion crises (VOC) in SCD, including the barriers and challenges patients face in managing their VOC at home. Speakers in this session included a representative from a patient advocacy group (Mr James, OBE) and two physicians, Dr Osunkwo and Dr Colombatti, who highlighted the importance of considering multiple perspectives when seeking to understand SCD. The viewpoints of healthcare professionals and patient advocates are important, but central to an understanding of how the disease impacts daily living is the perspective of patients themselves.
SICKLE CELL DISEASE: UPDATE ON EMERGING CHRONIC COMPLICATIONS
Myocardial Fibrosis and Diastolic Dysfunction: The Significance of Cardiac Complications
Doctor Punam Malik
Although VOC are the hallmark of SCD, a lack of VOC events does not prevent chronic multi-organ damage. Patients with sickle cell anaemia (SCA) have a median life expectancy of 45 years and this has remained unchanged for >30 years.1,2 Renal dysfunction is known to be a chronic comorbid condition in patients, as well as a mortality risk factor, and impairment of renal function during or after VOC, and independently of VOC, is often described with proteinuria, glomerulosclerosis, and tubular atrophy.3,4
Cardiopulmonary complications account for 40% of mortality in adults with SCD, and this mortality rate includes sudden death (6%), cardiac death (6%), and possible cardiac death (29%).2,5 In autopsy studies, sudden unexplained death in individuals with SCD occurred in 20–40% of patients, and these figures taken together suggest that cardiac complications are likely the main cause of adult mortality in this disease.6 The hyperdynamic physiology of patients with SCD is well known, and is secondary to anaemia. As a consequence of anaemia, patients have increased stroke volume and cardiac output, higher levels of chamber dilation, and normal-to-hyperdynamic systolic function. Atherosclerosis and myocardial haemosiderosis are uncommon in SCD, and the aetiology of any diastolic dysfunction is unclear.
A landmark study by Gladwin et al.7 showed a significantly higher mortality rate in patients with a tricuspid regurgitant velocity (TRV) of ≥2.5 m/sec, the majority of whom showed a modest increase in estimated peak pulmonary artery pressures of >25 mmHg systolic pressure; 30% of adults with SCD in the study were found to have pulmonary hypertension based on this definition. The Cincinnati Echocardiography Study of children8 (N=134; mean age: 12.3±4.7 years), supported by a meta-analysis of cardiac data in SCD, found that the most significant abnormality was left atrial volume, which was enlarged in 76% of patients. Older children also showed some evidence of early diastolic dysfunction.
It appears that patients with SCD develop a unique cardiac pathophysiology that is initially characterised by a high-output state with an increased cardiac output, normal-to-increased systolic function, and left ventricle dilation and enlargement, all of which occur secondary to chronic anaemia; in parallel, the patients develop a restrictive cardiac physiology that results in diastolic dysfunction and heart failure with preserved systolic function. Although this is not a restrictive cardiomyopathy in the classic sense, these patients have impaired diastolic filling and increased left atrial size and volume. In a study of 134 children with SCD (median age: 11 years), findings revealed a restrictive pattern of diastolic function in this patient population.9 Results showed that patients had severely abnormal echocardiographic measurements (E ratio: 18%; E/A ratio: 4%; lateral E/e’ ratio: 8%; septal E/e’ ratio: 14%). A meta-analysis of cardiac studies in SCD was conducted as part of the same study, concluding that a unique cardiomyopathy with restrictive physiology exists in patients with SCD.9 In children, there is often a progressive clinical course despite treatment, and these patients need careful monitoring with the consideration of cardiac transplantation.10 The impaired diastolic filling is thought to be a direct result of diffuse myocardial fibrosis, which prevents the heart from completely relaxing in diastole.11 The resulting back pressure onto the left atrium increases its size and volume, leading to further back pressure and modest increases in pulmonary venous hypertension with an elevation of TRV.8
In murine studies, it was found that diffuse microscopic myocardial fibrosis, detected by both histopathology and cardiac magnetic resonance (CMR) with myocardial T1 to quantify extracellular volume fraction (ECV), predisposes the myocardium to dysrhythmia and sudden death;11 this may explain the high rates of sudden death in SCD, although further studies are warranted. Another study of children, adolescents, and adults with TRV values of <2.5 (normal) or >2.5 revealed that every patient, regardless of age and TRV status, had evidence of significantly increased myocardial fibrosis (detected by CMR-based ECV quantification), compared with normal controls.9 Patients with SCD, therefore, have a hyperdynamic-restrictive cardiomyopathy which occurs secondary to diffuse microscopic fibrosis of the heart. In mice, diffuse myocardial fibrosis was seen by histologic analysis and was quantifiable by CMR imaging and gadolinium-based ECV quantification. In the human study, CMR-based ECV was performed prospectively.
Hydroxyurea: Fertility Is Still an Open Question
Doctor David Rees
Hydroxyurea is beneficial to nearly all patients with SCA, both in the short and long term and in most healthcare settings.12 Beneficial effects include reduced frequency of acute pain, acute chest syndrome, hospital admissions, and blood transfusions. It may also protect against progressive organ damage including nephropathy.13 The drug is undoubtedly underused in the majority of countries, although there is legitimate concern regarding the effects on fertility as it is a cytotoxic drug and affects dividing cells.14,15 It is recommended in national guidelines in many countries, including the UK and the USA, that all children with SCA should be offered hydroxyurea from a very early age. Although it is believed that treatment is unlikely to increase the risk of infertility in adulthood, there is not much evidence as yet to support this theory.12,16 There is no clear evidence of reduced fertility in females taking hydroxyurea, but it should be avoided in pregnancy because of the risk of teratogenicity unless there are very clear life-saving indications for hydroxyurea and in instances where no alternatives are possible.17 In males with SCD, there is a well-demonstrated reduction in sperm count even without hydroxyurea (40% of males with SCD have abnormally low sperm counts), and the observed reduction in sperm count that occurs on taking hydroxyurea is reversible in most cases.18 Therefore, infertility as a result of hydroxyurea is likely to be uncommon but, as a protective measure, sperm cryopreservation is justified in males before starting hydroxyurea treatment. Recent studies suggest that starting hydroxyurea in young children does not increase the risk of infertility, although there is relatively little research in this area.19 There is a need for more formal research on the effects on hydroxyurea on fertility in SCD with prospective studies. With improved survival in SCD, patient issues such as fertility become increasingly important.
Delayed Haemolytic Transfusion Reaction in Sickle Cell Disease: Prevention, Diagnosis, and Clinical Management
Doctor France Pirenne
Dr Pirenne explained how severe cases of delayed haemolytic transfusion reaction (DHTR) in patients with SCD are linked to hyperhaemolysis, a cause of death in 4% of patients with SCD. In hyperhaemolysis, destruction of both transfused and autologous red blood cells (RBC) occurs, and it is vital that these patients are recognised, diagnosed, and treated. Patients with SCD are at risk of DHTR because of their predisposition to making alloantibodies against transfused RBC.20 Clinical presentation is often comparable with a VOC, which is why DHTR is under-recognised. Diagnosis of DHTR is based on alloantibody screening and evidence of a rapid drop in adult haemoglobin concentration during any SCD exacerbation. Classical severe presentation includes acute bone pain, dark urine, profound anaemia, or increased lactate dehydrogenase, and these symptoms occur within 21 days post-transfusion.21 Mechanisms involved in hyperhaemolysis remain elusive but it is likely that complement is involved, as is toxicity of RBC content for vessel walls and inflammation.22
The main cause of DHTR is alloimmunisation against RBC antigens because of the high polymorphism of the blood groups involved (36 blood groups, 360 different antigens), depending on the ethnicity of an individual. Prevention of alloimmunisation is foremost, and the initial goal would be to match patients for all blood groups; however, a more feasible and pragmatic approach is to first match for RH/K groups. Once immunised, patients can be matched more extensively to FY, JK, and MNS groups. However, there are challenges present as a result of insufficient resources such as blood from donors of African ancestry, and concern regarding blood supply because of the lockdown during the COVID-19 pandemic. Furthermore, it is not possible to predict a patient’s transfusion risk if they have no historical transfusion data. Immunosuppressant therapy (rituximab, bortezomib) can be used for high-risk patients but only where there is an increased risk of developing new antibodies and in those with a history of DHTR.20,23,24
TREATMENT FOR SICKLE CELL DISEASE
Iron Overload Monitoring and Management
Doctor Ali Taher
Iron overload is an increasingly recognised problem, which may lead to increased morbidity and mortality in patients with SCD receiving long-term or recurrent transfusions. Thus, adequate and appropriate assessment tools for iron overload should be utilised to adequately assess the patient and implement treatment, such as liver iron concentration, transfusion history, and the importance of serial serum ferritin determination.25,26 The result of such assessments may indicate a need for chelation therapy, the key aim of which is to prevent long-term liver damage (cirrhosis) and cardiac iron overload in patients with SCD. However, the risk of extrahepatic damage is not absent, and the purpose of iron chelation therapy is to remove excess iron present in the organs, especially the liver, heart, and endocrine glands. Chelation therapy should be started after lifetime transfusions of 10–20 units, when serum ferritin is >1,000 ng/mL, or when liver iron concentration is >7 mg/g dry weight iron deposition (125 mmol/g), but should not be started in children under 2 years of age.26,27 Dr Taher suggested lower thresholds may be appropriate.
Deferasirox is an effective and well-tolerated chelator for the management of iron overload in patients with SCD. Results from a long-term study have shown that patients who received deferasirox for ≥4 years had a median serum ferritin significantly decreased by 591 µg/L (p=0.027; n=67).28 Monitoring urine and kidney function is essential to prevent severe renal damage, and is particularly recommended in patients under iron chelation therapy with deferasirox or desferrioxamine with nephrotoxic potential.29 A film-coated tablet formulation of deferasirox is available to improve adherence to iron chelation therapy through properties including improved palatability compared with a dispersible tablet taken as an oral suspension.
Sickle Cell Disease: Novel Treatment Options
Doctor Subarna Chakravorty
Dr Chakravorty described the complex pathophysiology identified in SCD and the therapeutic targets of various drugs available to treat patients. Pharmaceutical approaches to SCD therapy correlate with four broad pathophysiological ‘streams’ identified as potential therapeutic targets, and Phase I–III trials are underway to investigate new drug candidates (Table 1). L-glutamine has already received U.S. Food and Drug Administration (FDA) approval for use in SCD, and newer molecules such as canakinumab, montelukast, and simvastatin are in Phase II trials.30 Voxelotor, which acts by changing allosteric configuration of the sickle haemoglobin molecule and reducing intra-erythrocytic sickling, is the most recent drug approved by the FDA.
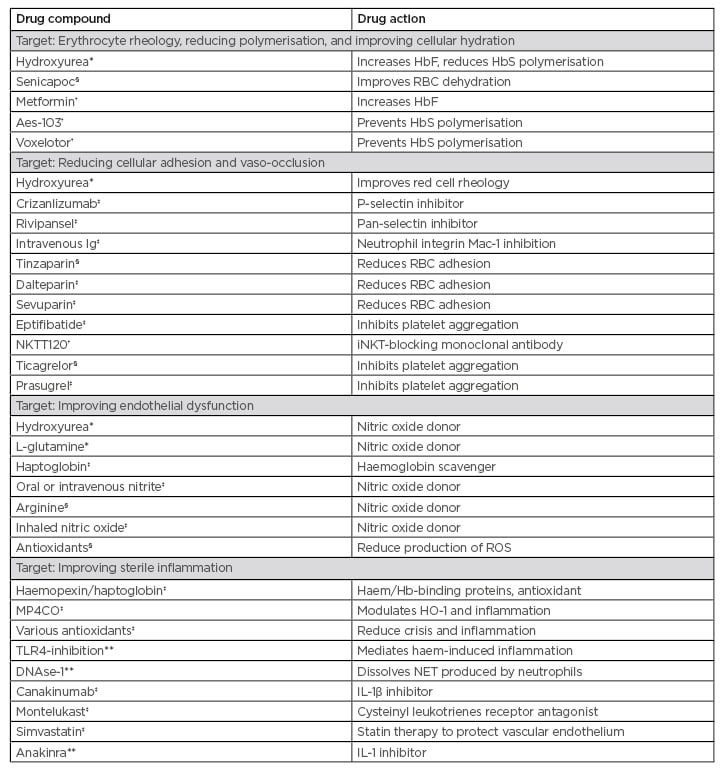
Table 1: Currently approved drugs and study trials of potential future treatments for sickle cell disease.
*U.S. Food and Drug Administration (FDA) approved for sickle cell disease
†Phase I RCT
‡Phase II RCT
§Phase III RCT
**Under investigation
Aes-103: 5-hydroxymethyl furfural; Hb: haemoglobin; HbF: fetal haemoglobin; HbS: sickle haemoglobin; HO-1: haem-oxygenase-1; iNKT: invariant natural killer T cell; NET: neutrophil extracellular traps; NKTT120: investigational therapy being developed by NKT Therapeutics; RBC: red blood cell; RCT: randomised controlled trial; ROS: reactive oxygen species; TLR4: toll-like receptor 4.
Haemopoietic stem cell transplants have been reported since 1984 in patients with SCD, with positive results. Gene therapy strategies in SCD, through gene addition, gene correction, or fetal haemoglobin induction, can address the lack of donor availability for haemopoietic stem cell transplant.31 There are 11 active trials in the USA and Europe which will likely revolutionise clinical practice, particularly if the cost can be reduced. The short- and long-term tolerability of the chemotherapy used in gene therapy is also an open question. These expensive trials are not without challenges when it comes to scalability, but if the transfection procedure can be simplified and made less expensive it could change how complicated SCD is managed in the future.
Recently, a Phase I study established clinical proof-of-concept for mitapivat, a first-in-class pyruvate kinase-R activator in SCD. Mitapivat may reduce sickle haemoglobin polymerisation and RBC sickling through a mechanism of decreased 2,3-diphosphoglycerate and increased ATP.32 Other novel interventions in SCD include red cell alternatives that act as O2 carriers;33 these nanoparticles may provide a treatment not only for SCD, but for other blood disorders which benefit from artificial haemoglobin. There are also advances in pain management, providing more choice for both physician and patient. Drugs such as cannabinoids, memantine, gabapentin, and others are also being investigated for management of pain.34 Regarding future treatment models, new drugs are increasingly being approved for use in patients and it is important to consider both selection and funding of the right drugs for patients. A polypharmacy approach, targeting multiple pathologies for a synergistic effect, may be a possible methodology in the future.
The ideal situation would be for an accessible gene therapy programme delivered in an ambulatory model of care, universal neonatal screening, and early therapeutic intervention. Empowerment of families to avail themselves of a reproductive choice, resulting in a reduction of the number of neonatal cases, and provision of a better service to countries which have higher numbers of cases such as Africa, India, and the Arabian Peninsula, are needed.
Hydroxyurea in Africa
Doctor Léon Tshilolo
Dr Tshilolo described how hydroxyurea is an attractive drug to treat SCD because of its many clinical benefits, low cost, and ease of oral administration. However, it is not used to a great extent in Africa, where 75% of affected children are born. In this setting, there are important coexisting conditions, such as endemic malaria and widespread malnutrition, which may affect the benefits of hydroxyurea.
There is also a presumptive fear among patients in Africa of sterility associated with the drug, leading to reduced use.
The REACH study evaluated the efficacy and safety of treating 600 children aged 1–10 years with hydroxyurea.35 Findings revealed that it is a feasible, safe, and effective treatment for African children with SCA including children aged <1 year, and may also be associated with reduced rates of malaria and other infections, leading to improved survival. Clinical benefits of reductions in all sickle-related events, of vaso-occlusive pain and acute chest syndrome, were sustained through the 36-month study period. These results indicated that hydroxyurea is a feasible, safe, and effective treatment for African children with SCA, and suggest that wider access to hydroxyurea for SCA has the potential to save millions of lives in Africa. It was also noted that many REACH participants had splenomegaly at the time of enrollment and many more developed the condition during the study. The condition is not a contraindication to starting hydroxyurea therapy in sub-Saharan Africa as, even with a large spleen, hydroxyurea is well tolerated and a beneficial dose can be achieved with excellent treatment responses, although the risk of malaria is increased in patients with splenomegaly.
MULTIPLE PERSPECTIVES ON THE BURDEN AND MANAGEMENT OF SICKLE CELL DISEASE
Effective Management of Vaso-Occlusive Crises: Overcoming Barriers and Finding Solutions
Doctor Ifeyinwa Osunkwo
Dr Osunkwo discussed how data from SWAY (3rd April 2019 to 4th October 2019),36 a worldwide patient and provider survey, looked at the impact of SCD on affected patients in various countries from several perspectives. It included >2,100 patients and 350 healthcare providers, and provided invaluable information from the patient’s perspective of how individuals cope with SCD and VOC. VOC are identified in many studies through patient utilisation (attendance) data from medical facilities, but this method can be limiting and often results in underestimated prevalence rates. Other studies, such as SWAY,36 an international, multi-country, cross-sectional survey which assessed the impact of SCD on patients’ daily lives, have used self-reported data to describe the epidemiology of VOC in SCD. SWAY provided a global insight into how patients managed SCD-associated pain, particularly how VOC were managed at home.
The survey showed that up to 95% of VOC have a short-term impact with time off from school or employment. In the 12-month period before the SWAY survey was completed, the mean prevalence of VOC was significant, at 5.3 per patient (standard deviation: 6.8), and 91% of patients experienced at least one VOC in the previous year.37 The survey also revealed geographical variation in the prevalence of VOC, with higher rates in the USA, Lebanon, and Bahrain, although reasons are unclear.38
Several studies showed that levels of patient pain are underestimated if they are only measured via healthcare facilities, indicating a need to improve patients’ experiences of acute care.39-41 Approximately 25% of VOC reported in SWAY were managed by patients at home.37 From the patient perspective, this was largely, but not exclusively, because of a perceived poor medical experience at the emergency room (ER) or hospital, which then prevented the patients from seeking further medical assistance when needed.42 In addition to the overcrowded and stressful hospital/ER environment, barriers to VOC management include poor transition from paediatric to adult care, the stigma associated with having SCD, ethnicity, and requiring opioids for pain control.43,44 Patients with SCD also wait longer to see an ER physician than, for example, a patient with a long bone fracture, which may be because of a lack of experience with SCD, or a lack of empathy as a result of the aforementioned stigma.45 The effects of medication on fertility rates and pregnancy are also of concern to patients. No clear evidence exists of a negative effect of hydroxyurea therapy on fertility, but further research is needed as more treated patients reach adulthood.13
Optimum management of VOC can be facilitated by pain action plans designed to educate patients on strategies to prevent triggers, advise on self-care strategies to manage pain at home, and inform patients when to seek medical assistance. Pain action plans also encompass guidance for acute pain management, discharge planning to prevent rebound-pain exacerbation, and co-ordination of care after discharge.46 This preventative approach helps patients to recognise their pain severity and manage their VOC accordingly (Figure 1).
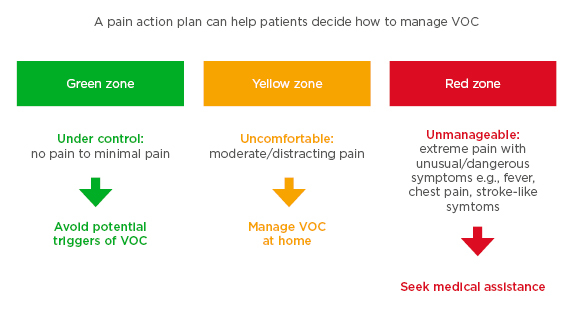
Figure 1: Example of a pain action plan for patients using traffic light colour-coding to highlight pain severity, showing the recommended treatment path according to pain level.
VOC: vaso-occuslive crises.
Impact and Treatment of Sickle Cell Disease: Hearing the Patient Viewpoint
Mister John James, OBE
Through the voices of patients, it is known that SCD can have a devastating impact on all aspects of patients’ lives, from education and employment to relationships and social interaction. Using the 7-point Likert scale,37 SWAY survey data demonstrated the profound effect of SCD on quality of life for patients worldwide, which is the most common treatment goal for patients with SCD.42 Two different studies of school children were carried out in the survey. In one group (<18 years of age, n=769), 46% reported that they often missed school. In the second group (>18 years of age, n=1,376), 51% reported that SCD had impacted their achievements at school, and 41% experienced decreased motivation at school.37 For the employed population there was a similar story; one-third of the patients reported that they had lost employment because of their disease, and 54% of the patients believed that they would have a higher income if they did not have SCD.37 In addition to limited therapeutic options, patients also have concerns about the long-term effects of treatments they receive, and of the perception and prejudgement of patients who need opioids and other strong pain-relief treatment, which relates to the stigma discussed previously.42 From the perspective of the patient advocacy group, there is a need to improve general awareness and understanding of SCD to challenge stigma and inequality, and to develop new treatment options to improve quality of life.
Living with SCD: Understanding the Consequences
Doctor Raffaella Colombatti
Dr Colombatti described how data from SWAY showed that the most commonly experienced SCD complications were fever, joint issues, and infection.37
As infectious disease still represents a major challenge in quality of life and survival for patients with SCD, antibiotic prophylaxis and global surveillance should be encouraged. There is a gap between the amount of emotional support patients actually need and how healthcare providers perceive the psychological burden of SCD, as patients’ perspectives of the clinical and emotional burden are often under-reported.47-50 Clinicians need to remember that SCD is a life-long chronic condition, where patients not only have physical complications that need to be treated, but are also affected by a profoundly life-changing disease. Chronic illnesses are associated with increased levels of loneliness and depression, which act synergistically to reduce overall wellbeing.47,51,52
REAL-TIME NATIONAL SURVEY OF COVID-19 IN HAEMOGLOBINOPATHY AND PATIENTS WITH RARE, INHERITED ANAEMIA
Doctor Paul Telfer
A late-breaking abstract from this virtual conference provided an update on incidence and mortality related to COVID-19.53 A national survey of patients with haemoglobinopathy and rare anaemia in England, UK, was conducted between 8th April and 6th May 2020 to provide clinical guidance and inform public health policy. The survey evaluated the impact of ‘shielding’ measures implemented by the UK government in March 2020 and included patients with all SCD subtypes. Findings revealed that a considerable number of patients with a haemoglobinopathy were infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) despite the advice to self-isolate at home. In children, the low incidence rate reflected the largely mild symptoms generally seen in this age group and, similarly, higher mortality rates among patients with SCD were observed with increasing age, as echoed in the general population. An unexpected finding from the survey was the lack of correlation between mortality as a result of COVID-19 and sex, or with clinical severity of comorbidities. Therefore, any decision to revoke shielding precautions in vulnerable patients requires a cautious approach.
FUTURE DIRECTIONS
The value of prevention in SCD is important in several contexts. Preconception screening of couples to avoid high-risk pregnancies is recommended, and females need relevant information on how to manage their pregnancy for better outcomes.54 The prevalence of ischaemic stroke by the age of 20 years is ≥11%, with the highest stroke rates occurring in early childhood.55,56 Transcranial Doppler ultrasound is used to identify patients at highest risk who may benefit from transfusion therapy.56-58 Prophylactic erythrocytapheresis addresses the problem of slow RBC transfusion, reducing the concentration of sickle haemoglobin-containing red cells, thereby improving symptoms of crises quickly.59 For the prevention of infection in children with SCD, prophylactic antibiotics initiated as early as 3 months of age is recommended.60
SUMMARY
SCD is a global disease; it is no longer only a disease of developing countries but is becoming more widespread in Europe and across the world. Earlier this year, the world experienced the devastating spread of SARS-CoV-2; for healthcare providers, this presented unprecedented challenges in managing care for patients with SCD. There are serious unmet needs to address if patients are to be treated successfully, especially because the pathophysiology of SCD is extremely complex, making it difficult to find a unique treatment. Barriers to the management of VOC include challenges in the ER, stigmatisation of patients with SCD, and poor transition from paediatric to adult care. Strategies to overcome these barriers include patient plans, day hospitals, and structured transition programmes. In this decade of new drug development, there is a need to examine the potential role of combination therapies. Hydroxyurea is a well-established drug and most trials are based on patients already on this treatment; adding another drug may become the standard of care in the future. However, because of the global distribution of patients and especially in developing countries, there is a need to consider the cost of new therapies. Many people do not have access to safe blood transfusions, therapeutic options, or even iron chelation and, to address the unmet needs in SCD worldwide, global collaboration is needed to develop treatments and protocols.





