
Presenters: Karen Yee,1 Shahram Kordasti2,3
1. Division of Medical Oncology & Hematology, Princess Margaret Cancer Centre, Toronto, Canada
2. Haematology Department, Guy’s Hospital, London, UK
3. Comprehensive Cancer Centre, King’s College London, London, UK
Disclosure: Yee reports honoraria and research funding from AbbVie, Astex, Forma Therapeutics, F. Hoffmann-La Roche, Genentech, Geron, Janssen, Jazz Pharmaceuticals, MedImmune, Novartis, Onconova Therapeutics, Inc., and Tolero Pharmaceuticals, Inc.; and had consultancy with Astex, Bristol Myers Squibb/Celgene, F. Hoffmann-La Roche, Novartis, Otsuka Pharmaceuticals, Paladin Labs, Pfizer, Shattuck Labs, Taiho Pharma, and Takeda. Kordasti reports research support from Celgene and Novartis; is a consultant for Boston Biomedical, Inc., API, and Alexion; speakers bureau for Alexion; and advisory board for Novartis.
Acknowledgements: This report was written by Jenny Lloyd, Compass Medical Communications Ltd., UK.
Support: The publication of this article was funded by Novartis. The views and opinions expressed are those of the authors and not those of Novartis.
Citation: EMJ Hematol. 2022;10[Suppl 4]:2-10.
Meeting Summary
Azacitidine and decitabine are established treatments for patients with higher-risk myelodysplastic syndromes (MDS). However, clinical responses may be suboptimal. The progression of higher-risk MDS is partly driven by factors that suppress the immune response, leading to ‘immune escape’ by leukaemic cells. Therefore, treatments that stimulate the immune response may benefit patients with higher-risk MDS. Shahram Kordasti presented an overview of immune dysregulation in MDS and its impact on disease progression. He then described current immunomodulatory treatments, including haematopoietic stem cell transplantation (HSCT) and immune checkpoint inhibitors, such as nivolumab, a therapeutic monoclonal antibody to the programmed cell death protein 1 (PD-1) receptor. However, the effectiveness of immune checkpoint inhibitors in MDS and myeloid leukaemia remains limited. Karen Yee then reported data on new drugs targeting the immune system. Magrolimab is a humanised anti-cluster of differentiation (CD) 47 monoclonal antibody with an overall response rate (ORR) of 91%, and a complete remission (CR) rate (CRR) of 42% when combined with azacitidine. Sabatolimab is an immuno-myeloid monoclonal antibody therapy to T cell immunoglobulin domain and mucin domain-3 (TIM-3), which is a promising therapeutic target in myeloid malignancies as it is expressed by leukaemic cells but not by normal haematopoietic stem cells (HSC). Sabatolimab activates macrophage and natural killer (NK) cell responses to leukaemic cells, with reported ORR and CRR of 58% and 20%, respectively, when combined with azacitidine or decitabine. Shahram Kordasti concluded the meeting by highlighting that, beyond the unmet clinical need for new immune-based therapies for higher-risk MDS, immune profiling of patients has an important role in comprehensive patient stratification for immuno-myeloid therapy.
Introduction
Karen Yee
The hypomethylating agents (HMA), azacitidine and decitabine, are currently regarded as the standard of care for patients with higher-risk MDS,1-3 but result in an ORR <50%, with a CRR <20%.4,5 Also, the mean duration of treatment response is reported to be 11–15 months,1,6 with a median time to develop acute myeloid leukaemia (AML) of <2 years.4,6 Patients who fail to respond to, or lose response to, therapy with HMAs have a median overall survival of <6 months.7,8 Therefore, there is a need for new treatment options for patients with higher-risk MDS who are not eligible for stem cell transplantation (SCT).
Dysregulation of the immune response contributes to the development and progression of MDS.9 Aberrant immune signalling and proinflammatory signalling pathways in a malignant myeloid cell clone and the bone marrow microenvironment are key players in the pathogenesis of MDS.9 Changes in the cytokine milieu lead to a switch from a proinflammatory state in lower-risk MDS to an immunosuppressive state in higher-risk MDS and AML.9 In lower-risk MDS, there is a decrease in regulatory T cells (Tregs),10 associated with targeting HSCs and myeloid progenitor cells by effector T cells.10 However, in the immunosuppressive stage higher-risk MDS, there is an expansion of myeloid-derived suppressor cells and Tregs, which leads to T cell exhaustion,11-16 suppression of anti-tumour responses,13,15-17 and development of immune tolerance12 and/or inhibition,14 resulting in immune evasion by the malignant cell clone. This dysregulated immune response provides a rationale for developing a targeted therapy in higher-risk MDS directed to oncogenic immune pathways.
The Role of the Immune System in Higher-Risk MDS
Shahram Kordasti
There is evidence that conditions associated with chronic immune stimulation can activate toll-like receptor (TLR)-mediated immune responses, initiate proinflammatory events to induce a ‘myeloid bias’ of myeloid stem and progenitor cells, and result in the production of proinflammatory cytokines, reactive oxygen species, and damage-associated molecular patterns (DAMP [ Figure 1 ]).9 This ‘smouldering inflammation’ is a driver of MDS, contributing to additional genomic instability and somatic mutations.9 As MDS progresses, the number of myeloid cell mutations increases to drive the progression to higher-risk MDS, and malignant transformation to AML.
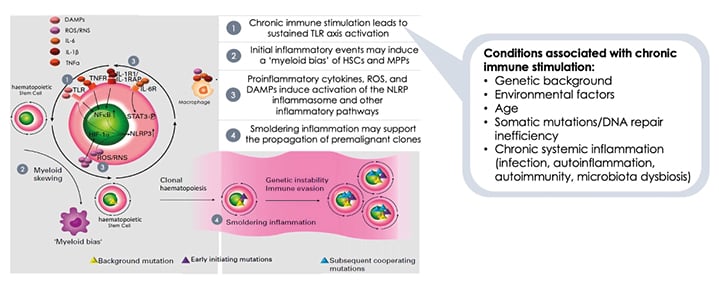
Figure 1: Immune dysregulation contributes to the development of myelodysplastic syndromes.
DAMPs: damage-associated molecular patterns; HIF-1α: hypoxia-inducible factor subunit 1-alpha; NFκB: nuclear factor κB; NLRP3: nucleotide-binding oligomerisation-like-receptor family pyrin containing domain 3; RNS: reactive nitrogen species; ROS: reactive oxygen species; STAT3: signal transductor and activator of transcription 3; TLR: toll-like receptor; TNFR: TNF receptor.
Adapted from Winter S et al.9
MDS may be considered to represent part of a disease spectrum from an autoinflammatory to an autoimmune state, with shared genetic associations and inflammatory pathways connected by interactions between innate and adaptive immune responses.9 At any given time, patients may present or develop at a stage of this MDS disease spectrum.
Lower-risk MDS is associated with a proinflammatory immune response and activation of effector immune cells, which promote myeloid cell apoptosis.18 Patients with lower- and higher-risk MDS have increased levels of the activated nucleotide-binding domain and leucine-rich repeat pattern recognition receptor family, pyrin domain-containing protein 3 (NLRP3) inflammasomes and DAMPs.9 HSCs respond to proinflammatory signals and DAMPs via elevated TLR expression;9 and to DAMP signalling through TLRs, leading to nuclear factor κ B (NFκB) transcription of IL-1β and priming of the inflammasome.9 The primed NLRP3 inflammasome activates the expression of IL-1β, which leads to inflammatory cell death.9 The resulting release of proinflammatory cytokines, reactive oxygen species, and DAMPs results in a cycle of inflammation and cell death in the bone marrow (Figure 1).9
However, in patients with higher-risk MDS, the immune pathway is predominantly immunosuppressive,9 resulting in the suppression of the immune response that could keep malignant leukaemic cell clones under check. This response is associated with a significant expansion of immunosuppressive cells that include Tregs and myeloid-derived suppressor cells, and a reduction in the number and function of dendritic cells, CD8+ T cells, and NK cells.9 This progressive immune pathway includes the following stages: activation of CD8+ T cellsby malignant MDS stem cells, leading to the expansion of T cell populations; secretion of proinflammatory cytokines by T cells into the microenvironment of the bone marrow, such as TNFα and interferon γ, which results in inhibition of haematopoiesis and the induction of inhibitory molecules; and MDS stem cells may express inhibitory immune checkpoint receptors, resulting in escape from malignant cell surveillance, suppression of T cell proliferation, and induction of T cell exhaustion and apoptosis.19,20 Overall, this immune pathway transforms a very active inflammatory environment to one of ‘smouldering inflammation’ and immunosuppression.
In higher-risk MDS, suppression of multiple immune cell types leads to the phenomenon of malignant cell escape by leukaemic myeloid cells.10,21 Patients with higher-risk MDS have fewer functional/active CD8+ T cells but higher numbers and increased activity of Tregs, which promotes tumourigenesis through loss of immune surveillance.10,21 They also have decreased levels and activity of NK cells, and dendritic cell secretion of anti-inflammatory cytokines that induce Tregs.10,21 Therefore, in higher-risk MDS, immune cell dysfunction results in tumour escape, or malignant cell escape, by leukaemic myeloid cells.10,21
For patients with lower-risk MDS, immune-based treatment options could be used, which include immunosuppressive therapy (e.g., anti-thymocyte globulin) and immunomodulatory drugs (e.g., lenalidomide).9,22 As the immunosuppressive milieu in higher-risk MDS cannot eradicate the malignant cell clones, reactivating the immune system is a promising approach for higher-risk MDS. Treatment options include HSCT and donor lymphocyte infusion, which have a graft-versus-leukaemia effect9,22 that can reprogramme the immune system, potentially allowing for control over the malignant cell clone. HSCT can also reprogramme the immune dysregulation in MDS,9 while donor lymphocyte infusion has been shown (in AML) to downregulate PD-1 and TIM-3 and increase interferon γ and TNFα production.23 HMAs have a mixed effect on the immune system: decreasing regulatory function and the cytokine profile of Treg, but increasing the expression of immune checkpoints such as PD-1, programmed death-ligand 1, and cytotoxic T lymphocyte-associated protein 4 (CTLA-4).24-26
Immune checkpoints regulate immune homeostasis by balancing the signals that mediate the T cell immune response.27 When T cells are in an inflammatory environment, they can protect themselves from excessive proliferation and cell death by upregulation of immune checkpoints, including PD-1 and CTLA-4.28,29 Upregulation of inhibitory immune checkpoints in the microenvironment of MDS and leukaemic cells is essential for cell survival in myeloid malignancies.30 However, malignant cells may evade this system by inducing cell depletion and reducing the activity and function of T cells. While checkpoint blockade to revive the immune system works in some cancers, the traditional immune checkpoint inhibitors, including nivolumab, ipilimumab, pembrolizumab, and durvalumab, have limited survival benefits in patients with myeloid leukaemias and MDS.31-34
In MDS, somatic mutations contribute to immune suppression.35 T cells are chronically activated by leukaemia cells, leading to T cell exhaustion or depletion.36 In solid tumours, T cells are confined within the neoplastic cell microenvironment, and checkpoint blockade can enable these confined T cells to recognise specific antigens and kill the cells that express these antigens. However, in leukaemias such as AML, the cells are exposed and the most immunogenic cells have likely already been eliminated. When checkpoint blockade is given, it is harder to activate antigen-specific T cells that target malignant clone(s).
TIM-3 is a key regulator of adaptive and innate immune responses in myeloid malignancies.14,17 Patients with TIM-3 loss-of-function mutations exhibit a severe autoimmune phenotype characterised by excessive production of proinflammatory cytokines, including IL-1β and IL-18.17 As TIM-3 is expressed on leukaemic stem cells but not normal HSCs,14,17,37,38 it may be possible to target cancer cells. There is also the potential to reactivate the immune system. The TIM-3 and galectin-9 loop drives the self-renewal of leukaemic stem cells by activating the NFκB pathway, and the secretion of TIM-3 and galectin-9 allows evasion of immune surveillance.17 TIM-3 is expressed on several immune cells, including T cells, NK cells, and macrophages,17 making it an attractive target in MDS and AML.
Overall, the immune response in MDS is complex, and stratified treatment approaches or effective combination therapies are lacking, especially in higher-risk MDS, where there is a high unmet need for effective treatments with durable responses.9 However, finding the best combination for each patient needs more investigation and more stratification. While the International Prognostic Scoring System (IPSS) is excellent for predicting the outcome of patients with MDS, it does not take immune factors into account, so different approaches will be required to stratify patients.
Immune-Based Treatments in Development for Higher-Risk MDS
Karen Yee
HMAs have received regulatory approval for the treatment of patients with higher-risk MDS. However, real-world data indicate limited survival benefits39-41 and significant adverse events.42 Therefore, there is a need for improved therapies that improve outcomes and have safety profiles that are better than, or at least similar to, currently approved HMAs.
Several new treatment approaches are in development to optimise clinical outcomes in patients with higher-risk MDS. Some of these therapies target immune-based pathways, including PD-1 (nivolumab, pembrolizumab), CTLA-4 (ipilimumab), TIM-3 (sabatolimab), and CD47 (magrolimab).43 Others target B cell lymphoma-2 (venetoclax); neural precursor cell expressed, developmentally downregulated protein 8 (pevonedistat); isocitrate dehydrogenase-1 (enasidenib); isocitrate dehydrogenase-2 (ivosidenib); FMS-like tyrosine kinase 3 (midostaurin, gilteritinib); and TP53 (eprenetapopt).43 These new targeted therapies have been evaluated as single agents and in combination with an HMA, but emerging data have not been encouraging, underscoring the need for more options.
Magrolimab is a macrophage immune checkpoint inhibitor that targets CD47, a transmembrane protein that functions predominantly as a ‘don’t eat me’ signal.44 CD47 binds to signal regulatory protein α on macrophages and generates a signalling cascade that prevents phagocytic elimination by macrophages. CD47 is expressed on normal cells and overexpressed on myeloid malignancies and cancer cells. However, when CD47 is blocked, as with magrolimab, there is selective elimination of cancer cells that express both CD47 (‘don’t eat me’) and pro-phagocytic (‘eat me’) signals, as healthy cells do not express ‘eat me’ signals. However, when normal cells become damaged (by stress responses, DNA damage, atypical cell division, etc.) or are ageing red blood cells, they express pro-phagocytic signals that lead to their removal by phagocytosis (e.g., programmed cell removal). Similarly, when CD47 is blocked in malignant cells, they still express an ‘eat me’ signal, facilitating phagocytosis by macrophages.
Efficacy data from a recent Phase Ib study have shown that magrolimab in combination with azacitidine as frontline treatment for patients with higher-risk MDS are encouraging.45 Among 33 efficacy-evaluable patients, the ORR was 91%, with a CRR of 42%.45 Also, 24% achieved a bone marrow CR (mCR), including four patients with haematological improvement (HI).45 Medium time to response was rapid (1.9 months).45 The treatment combination was well tolerated, with a similar safety profile to single-agent azacitidine.45 Among patients dependent on red cell transfusions, 58% achieved transfusion independence.45 Among patients with abnormal cytogenetics, 35% achieved a complete cytogenetic response.45 Currently, a Phase III randomised study of magrolimab plus azacitidine versus placebo plus azacitidine in patients with treatment-naïve, higher-risk MDS is underway (NCT04313881).46
Sabatolimab is a novel humanised therapeutic monoclonal antibody that targets TIM-3. The TIM-3 inhibitory receptor is involved in regulating both innate and adaptive immune responses.47,48 It is expressed on immune cells and is also aberrantly expressed on leukaemic stem cells, but not on normal HSCs.47,49-51 Leukaemic cells can also secrete galectin-9, a ligand of TIM-3. Binding of galectin-9 by TIM-3 leads to upregulation of NFκB signalling, as well as β catenin signalling, which results in leukaemic stem cell self-renewal (Figure 2A).38,47,52 This autocrine stimulatory loop is essential for leukaemic stem cell proliferation. It has been postulated that when sabatolimab binds to TIM-3 on immune cells, this activates macrophages and NK cells to attack leukaemic stem cells and blasts (Figure 2B), leading to cell phagocytosis. Furthermore, sabatolimab interferes with the interaction between galectin-9 and TIM-3, inhibiting the autocrine stimulatory loop and impeding leukaemic cell proliferation.
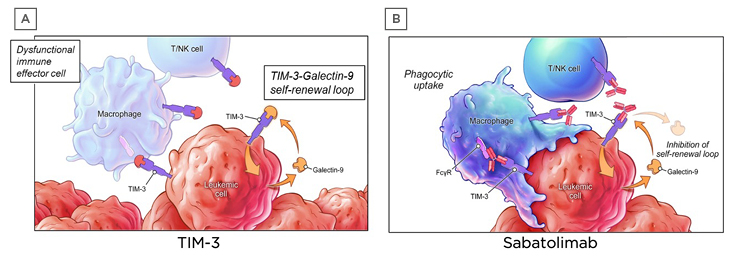
Figure 2: Sabatolimab targets T cell immunoglobulin domain and mucin domain-3 on immune and leukaemic cells.
FcγR: Fc gamma receptor; NK: natural killer; TIM-3: T-cell immunoglobulin domain and mucin domain-3.
In a Phase Ib study, sabatolimab was given with either azacitidine or decitabine to 53 patients with higher-risk MDS and 48 patients with AML.53 The safety profile of the combination was generally similar to that of single-agent HMA.53 Only four patients (4%) experienced a treatment-related, immune-mediated adverse event that led to discontinuation, reduction, or interruption of sabatolimab.53 There were no grade ≥3 treatment-related, immune-mediated adverse events in patients with MDS.53 Among 50 response-evaluable patients with higher-risk MDS, the ORR was 58%, with a CRR of 20% and an mCR rate of 24%, including 10% with HI (Figure 3).53 The median duration of response was 16.1 months, increasing to 21.5 months among those with a CR.53 The estimated 1-year progression-free survival rate was 50%, and 23% of patients proceeded to SCT.53
Subgroup analyses of patients with adverse genetic features, including TP53 mutation, ≥1 of TP53/RUNX1/ASXL1, and poor or very poor cytogenetic risk, were performed. Among 14 patients with a TP53 mutation, the ORR was 71%, with a CRR of 29% and an mCR with HI in 14% of patients (Figure 3).53 The responses were durable, with a median duration of response of 14.7 months. Results among 31 patients with TP53, RUNX1, or ASXL1 mutations were similar (Figure 3).53 Among those with very poor-risk cytogenetics, although the ORR was 65%, with a CRR of 25%, the median duration of response was only 7.9 months (Figure 3).53 Phase II (NCT03946670)54 and III (NCT04266301)55 studies comparing sabatolimab plus HMA versus placebo plus HMA in patients with treatment-naïve higher-risk MDS are underway.
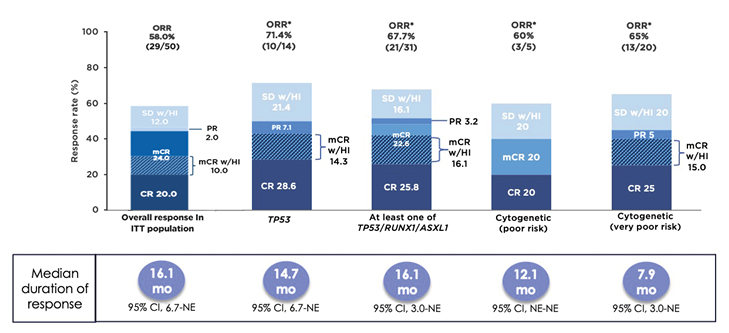
Figure 3: Patients with higher-risk myelodysplastic syndromes were able to achieve durable responses when treated with sabatolimab plus hypomethylating agent.53
*Evaluable patients, including patients with a valid baseline and ≥1 post-baseline bone marrow assessment, or if they had disease progression or disease-related death prior to the first marrow assessment.
CI: confidence interval; CR: complete remission; mCR: bone marrow complete remission; mCR w/HI: bone marrow complete remission with haematologic improvement; NE: not estimable; ORR: overall response rate; PR: partial remission; SD w/HI: stable disease with haematologic improvement.
CA-4948 is an oral small-molecule inhibitor of IL-1 receptor-associated kinase (IRAK)4, which is a downstream effector of the TLR and IL-1 receptor pathways.56,57 These pathways are often dysregulated in MDS and AML.56 TLR stimulation leads to recruitment of the adapter protein, myeloid differentiation factor 88, which binds to IRAK4 and activates IRAK1, leading to activation of NFκB.57 Overactivity of NFκB leads to inflammation of the leukaemic cell microenvironment, suppression of apoptosis, malignant cell survival, and cell proliferation.57 The oncogenic long form of IRAK, which is generated by alternative splicing in patients with MDS and AML and leads to maximal activation of NFκB, is preferentially expressed in >50% of patients with AML.58 Activated IRAK4 has been identified as a driver of adaptive resistance in AML.59 CA-4948 has been shown to inhibit IRAK4, leading to reduced leukaemic cell proliferation.57
CA-4948 is currently under investigation in a Phase I dose-escalation study of 11 patients with higher-risk MDS, and 11 patients with AML.57 Among nine response-evaluable patients with higher-risk MDS, one achieved a CR, two achieved mCR, and four patients had stable disease.57 Reduction in marrow blasts was achieved at all dose levels and in 10 out of 12 patients with elevated blasts.57 Among patients with a significant narrow blast reduction, there were also signs of HI.57 CA-4948 was also effective in heavily pre-treated patients.57 Treatment is ongoing in six out of 17 evaluable patients, and one patient proceeded to an SCT.57 CA-4948 monotherapy was safe and tolerable at doses of 200 and 300 mg twice daily, but there were dose-limiting toxicities at 400 and 500 mg twice daily, including reversible rhabdomyolysis and syncope.60
Other emerging immune-based therapies include: bispecific T cell engager therapies, which link endogenous T cells to antigens expressed by leukaemic cells, thus activating the cytotoxic potential of patient T cells;61 chimeric antigen receptor T cell therapy, which involves synthetic constructs that enable recognition and killing of target cells in a highly specific and major histocompatibility complex-independent manner;62 and antibody–drug conjugates, which are monoclonal antibodies that are covalently bonded to a cytotoxic compound.63 Selective targeting of cell surface antigens by antibody-drug conjugates kills malignant cells by pinpointing delivery of cytotoxic drugs. NK cell-based immunotherapies include adoptive NK cell transfer, chimeric antigen receptor-NK cell therapy, antibodies targeting tumour-associated antigens, bispecific killer cell engager, tri-specific killer cell engager, NK cell inhibitory receptors, cytokines, and drugs with immunomodulatory function.64
In summary, there is an unmet need for new therapies that can provide improved outcomes and durable responses and have a good safety profile in patients with higher-risk MDS who are ineligible for HSCT. Given the broad role of the immune system in MDS disease development, multiple immune-based treatments are currently being explored, with some encouraging results.
Closing Remarks
Shahram Kordasti
The immune response in MDS is as complex as the disease itself and follows a stepwise progression from lower-risk to higher-risk MDS. The immune response changes as MDS progresses and involves a combination of changes in the inflammatory microenvironment, the types of immune cells, the cellular and humoral immune responses, and genomic instability. However, every MDS patient is different, with some having more inflammatory features and some having more autoimmune features. Therefore, it is important to tailor treatments to individual patients at each stage of their clinical course.
As patients with MDS often have an inflammatory response and autoimmune features, combination treatments may be beneficial to target and modulate both. Although the best combinations are currently unknown, ongoing trials of immune-based therapies may show the benefits of reviving the immune system. Beyond the unmet clinical need for new immune-based therapies, there is also a need for a more comprehensive approach for patient stratification, including immune profiling. This treatment approach could be based on clinical and multi-omics data on identified somatic mutations and targeted transcriptomics of inflammatory genes, the immunome, gut microbiome, and neoantigen profiling.9 Patients could be classified using autoinflammatory, autoimmune, immunogenic immunosuppressive, and non-immunogenic immunosuppressive profiles.9 Therefore, individualised, or personalised, targeted immuno-therapy in patients with higher-risk MDS, resulting in safe and durable responses, may be realised.





