
Presenters: Rachel Kesse-Adu,1 Erfan Nur,2 Kofi A. Anie,3 Robert Clark Brown4
1. Guy’s and St Thomas’ NHS Foundation Trust, London, UK
2. Amsterdam University Medical Centers, Academic Medical Centers, the Netherlands
3. London North West University Healthcare NHS Trust, Imperial College London, UK
4. Aflac Cancer and Blood Disorders Center, Children’s Pediatric Institute, Emory School of Medicine, Atlanta, Georgia, USA
Disclosure: According to the Code of Medical Ethics, Kesse-Adu, Nur, and Anie declare that their presentation is sponsored by the pharmaceutical company Novartis; they state that the content of the presentation represents his/her independent views, to contribute to the sharing of medical knowledge, and does not constitute advertising of medicinal products within the meaning of the Pharmaceutical Law and regulations issued thereunder. Brown reports membership on an advisory committee for Global Blood Therapeutics; consultancy for Imara, Novartis, and Novo Nordisk; and research funding from Global Blood Therapeutics, Forma Therapeutics, Imara, and Novartis.
Acknowledgements: This report was written by Jenny Lloyd, Compass Medical Communications Ltd., UK.
Support: The publication of this article was funded by Novartis. The views and opinions expressed are those of the authors and not necessarily those of Novartis.
Citation: EMJ Hematol. 2021;9[Suppl 6]:2-11.
Meeting Summary
Rachel Kesse-Adu highlighted that the hallmark of sickle cell disease (SCD) is painful vaso-occlusive crises (VOCs). These not only cause acute pain, but the underlying mechanisms can result in complications, including acute chest syndrome (ACS), avascular necrosis, acute splenic sequestration, priapism, and chronic organ damage. As a consequence, SCD is associated with high healthcare utilisation, morbidity, and mortality. Erfan Nur explained the importance of tailoring treatment to each patient with SCD. He discussed traditional therapies, such as blood transfusions, haematopoietic stem cell transplantation, and hydroxyurea, as well as the novel alternatives L-glutamine, crizanlizumab, and voxelotor. It was explained that gene therapies (gene addition, gene editing, and gene silencing) are a promising new potential treatment for patients with SCD, although these are in the early stages of development. Kofi A. Anie discussed the psychosocial problems associated with SCD and the high burden this condition has on the emotional wellbeing of patients. He went on to discuss psychological interventions, including cognitive behavioural therapy (CBT), mindfulness-based cognitive therapy, and mobile health apps. Robert Clark Brown highlighted a potential new tool, the Sickle Cell Pain Diary-Self Report (SCPD-S) app, which was developed to evaluate the impact of VOCs on school/work and capture the physical and emotional difficulties experienced by patients. The next speaker reported that, in a single centre in Brazil, 11% of patients with SCD had severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) antibodies. Seropositivity was significantly associated with older age and regular public transport use. Only one out of 135 patients with SCD was hospitalised with COVID-19. In the next presentation, it was reported that, among 11 patients with SCD and COVID-19 in Lebanon, most had mild-to-moderate COVID-19, but one adolescent developed multisystem inflammatory syndrome.
The Long-Term Impact of Sickle Cell Disease: Understanding the Role of Vaso-occlusive Crises in End Organ Damage
Rachel Kesse-Adu
The pathophysiology of SCD is complex.1-3 Sickled red blood cells (RBCs) can undergo haemolysis and cause vaso-occlusion. Together with various other processes (e.g., oxidative stress, nitric oxide depletion, complement activation, multicellular adhesion, chronic vascular inflammation, and ischaemia-reperfusion injury), this can result in VOCs, haemolytic anaemia, and, ultimately, end organ damage. Many different organ systems can be affected, with patients at risk of stroke, ACS, hepatopathy, cholelithiasis, avascular necrosis, retinopathy, heart disease, acute splenic sequestration, nephropathy, and priapism.2 Some of the most disabling consequences of SCD are acute stroke and chronic cerebral ischaemia.4 Cerebral infarction occurs in approximately 10% of patients by age 20 years,5 while silent cerebral infarctions (which occur in approximately 17% of paediatric patients) are linked with poor cognitive functioning and learning development.5
VOCs, which are severe pain crises, are the hallmark of SCD. VOCs have four distinct phases:6
1. Prodromal phase: numbness, paraesthesia, aches, or can be asymptomatic.
2. Initial phase: anxiety increases and pain severity peaks.
3. Established phase: pain severity remains at a maximum, signs of joint effusion and inflammation.
4. Resolving phase: pain severity decreases.
Varying definitions of a VOC have been used in trials.7-9 However, the current criteria outlined by the ACTTION-APS-AAPM Pain Taxonomy (AAAPT)10 for the diagnosis of acute SCD pain are increased pain for ≥2 hours that had started in the past 10 days; at least one of: palpation elicits focal pain/tenderness, movement elicits focal pain, or a decreased range of motion/weakness; and pain that is not entirely explained by specific physical examination findings or imaging abnormalities.11
Renal structure and function are also impacted by SCD.12 Acute kidney injury, which may be due to repeated cycles of hypoxia and ischaemia associated with VOCs,13 develops in approximately 75% of VOC episodes that are complicated by acute multiorgan failure, and haemodialysis is required in around 18% of these episodes.12 Approximately 30% of adult patients develop chronic renal failure, which can be fatal.2
ACS is characterised by fever and hypoxia requiring supplemental oxygen therapy, plus a new pulmonary infiltrate on chest X-ray.14 Recurrent episodes are a significant risk factor for sickle chronic lung disease and death.15 Infection, embolism, and pulmonary infarction all contribute to the initial pulmonary damage.16 Avascular necrosis occurs in up to 50% of patients with SCD by age 35 years,17 commonly in the femoral head.18 In a 40-year study of 1,056 patients, previous hospitalisation for VOCs was significantly associated with an increased rate of avascular necrosis.15
Acute splenic sequestration, characterised by an abrupt onset of pallor, weakness, tachycardia, abdominal pain, and distention,19 occurs in 10–30% of children with SCD, and is a leading cause of death. The trigger for splenic sequestration is often unknown, but it can occur alongside or shortly after a VOC or infection.20
Priapism, which is caused by vaso-occlusive obstruction of the venous drainage of the penis, affects nearly 90% of men with SCD.21 It can be a urological emergency, and lack of prompt treatment can result in erectile dysfunction.22 Data from the Cooperative Study of SCD reported that priapism was significantly associated with VOCs.23
Sickle hepatopathy includes liver dysfunction and hyperbilirubinaemia.24 Acute hepatic sequestration is a rare but severe complication of VOCs, occurring in approximately 10% of patients.25
Patients with SCD often incur damage to multiple organs.26 For example, pulmonary hypertension has been linked with nephropathy, markers of renal injury, cutaneous ulceration, and priapism.26 In a retrospective–prospective cohort study of 150 adults with SCD, 24% had multiple organ impairment (two or three of: lung, heart, kidney).26 Time to death was considerably shorter among those with two or three versus zero or one organs impaired (8 versus 14 years).26 Furthermore, patients with three versus zero affected organs were much more likely to die as a result (85.7% versus 8.2%).26
Although chronic organ damage results in morbidity and mortality among patients with SCD, it often goes unrecognised. In an autopsy study of patients with SCD, 75% had evidence of chronic organ damage at autopsy, but only 25% had chronic organ damage in their clinical histories.27
Fortunately, the life expectancy of patients with SCD has improved with better treatment and management strategies.28 However, a longer lifespan can be associated with repeated VOCs and accumulating organ damage.29 In a 7-year study, approximately 60% of adults with SCD developed a new SCD-related complication.30 Screening to minimise organ damage is therefore important, namely:
- annual screening of children aged 2–16 years with transcranial Doppler for stroke risk;31
- screening for retinopathy with a dilated eye examination from age 10 years;31
- early screening for hypertension and microalbuminuria to prevent or minimise renal failure;32 and
- echocardiography to screen for suspected pulmonary hypertension.33
Prevention of VOCs may reduce the risk of organ damage and subsequently reduce mortality rates. However, SCD-related end organ damage has been associated with a substantial economic burden in over 10,000 patients with SCD who are on Medicaid.34
Knowing the Options: Personalising Treatment of Sickle Cell Disease
Erfan Nur
There is a unique set of factors to consider in the treatment decision-making process for each patient with SCD, as detailed in Figure 1.
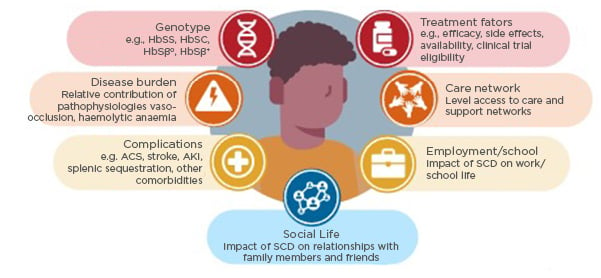
Figure 1: Factors to consider in the treatment decision process for a patient with sickle cell disease.
ACS: acute chest syndrome; AKI: acute kidney injury; SCD: sickle cell disease.
Blood transfusions (simple or exchange) can dramatically reduce the risk of stroke by 92% versus placebo among patients with transcranial Doppler velocity ≥200 cm/s in one trial.35 However, after a first stroke, patients with SCD can still experience progressive cerebral infarcts, silent strokes, or a second overt stroke while receiving transfusions.36 This risk is greatly increased among patients with cerebral vasculopathy.36 Acute transfusions may be beneficial for patients with ACS, acute splenic sequestration, or priapism.37 Chronic transfusions can be used in patients with cerebrovascular disease, select pregnancy complications, debilitating frequent pain, etc.38 However, potential complications include iron overload, RBC alloimmunisation, and delayed haemolytic reactions.37
Haematopoietic stem cell transplantation is the only potentially curative treatment option for patients with SCD. For patients without a matched sibling donor, haploidentical stem cell transplants are a promising alternative.39,40 Unfortunately, myeloablative conditioning can result in toxicity, such as in graft-versus-host disease. Complications can be reduced with non-myeloablative conditioning, but the graft failure rates are higher.41
Hydroxyurea, which is the mainstay of SCD therapy, increases fetal haemoglobin levels, thus inhibiting the polymerisation of haemoglobin S.42 In a double-blind trial, hydroxyurea versus placebo significantly reduced the rate of VOCs (median rate: 2.5 versus 4.5 per year), and increased the median times to the first (3.0 versus 1.5 months) and second (8.8 versus 4.6 months) crises.42 However, patient adherence can still be an issue.43
Recently, alternative treatment options have emerged, including:44
- L-glutamine, an amino acid that decreases oxidative stress and RBC adhesion, thus potentially reducing the frequency of VOCs.45,46 In a Phase III, placebo-controlled, randomised trial, L-glutamine significantly reduced VOC frequency (by 25%) and hospitalisations (by 33%) among patients aged ≥5 years with ≥2 VOCs in the past year.45
- Crizanlizumab, an anti-P-selectin monoclonal antibody that nearly halved the annual rate of VOCs when dosed at 5 mg/kg (median: 1.6 versus 3.0 with placebo; p=0.01) in a Phase II, randomised SUSTAIN study.9 It also increased the time to first VOC (4.1 versus 1.4 months; p=0.001).9
- Voxelotor, a haemoglobin S polymerisation inhibitor that increased haemoglobin levels at a dose of 1,500 mg versus placebo (+1.1 versus -0.1 g/dL; p<0.001) in a Phase III, randomised HOPE trial,47 with 51% versus 7% achieving an increase of >1 g/dL. The annual rate of VOCs was also lower (2.8 versus 3.2), although significance was not reported.47
Overall, when deciding on treatment for patients with SCD, factors to consider include disease-related factors (genotype, relative contribution of disease pathologies, and complications/comorbidities), patient-related factors (impact of SCD on school/employment, social life, and wellbeing; treatment preference; logistical issues; and care network), and treatment-related factors (efficacy, tolerability, and availability).
Gene Therapy for Sickle Cell Disease: Where Are We Today?
SCD arises from a single point mutation that causes a significant change in the structure of haemoglobin, resulting in abnormal haemoglobin polymerisation upon deoxygenation.16 Pain is the hallmark of SCD, but while pain is the visible outcome of VOCs, they also cause underlying adhesion, inflammation, activation, and endothelial damage, and therefore may potentially bring about acute and chronic complications, morbidity, and mortality.48
SCD gene therapy is based on autologous stem cells.49 Firstly, the patient’s own stem cells are harvested. Then, gene therapy techniques are used to introduce or increase the production of healthy γ- or β-globin genes from stem cells. These are then transplanted back into the patient. The benefits of this method are that the patients act as their own donor, so treatment is available to all patients; there is no need for immunosuppression; and there is no risk of graft-versus-host disease.
Gene addition therapy involves adding a new gene, by using a viral vector to deliver a non-sickling globin gene to the stem cells.50 This viral vector’s genes are removed and replaced with the anti-sickling gene. This does not remove or change any of the existing genes. The most well studied example of this therapy is LentiGlobin, which causes the production of haemoglobin AT87Q.51 Among 19 patients with a history of severe vaso-occlusive events and 6 months of follow-up, LentiGlobin resulted in complete resolution of vaso-occlusive events.51
Gene editing involves editing a gene in stem cells by removing, disrupting, or correcting faulty elements of the DNA. An enzyme is used to cut the DNA at one location, allowing the sequence to be changed with high precision. Current trials aim to reactivate protective fetal haemoglobin or directly correct the variant that causes the disease (gene correction). In a Phase I/II PRECIZN-1 trial,52 zinc finger nucleases were shown to bind and cleave specific DNA sequences in the genome, turning fetal haemoglobin back on to reduce symptoms of SCD. Similarly, clustered regularly interspaced palindromic repeats (CRISPR)-Cas9 gene editing technology can be used to correct disease-causing mutations.53 In the ongoing CLIMB study,54 CRISPR-Cas9 has been used to increase fetal haemoglobin in two patients with SCD, with early beneficial effects on VOCs.55
Gene silencing involves silencing the BCL11A gene that switches off fetal haemoglobin, thereby increasing (non-sickling) fetal haemoglobin and reducing (non-sickling) adult haemoglobin in the patient. In a small study, the robust, stable induction of fetal haemoglobin was seen in six patients.56
Gene editing/addition involves editing a gene that is in the body and adding an oligonucleotide strand to produce a new gene product. The homologous directed repair pathway uses a homologous DNA strand, which acts as a template for high-fidelity double-strand break repair.57
Overall, gene therapy efficacy is measured based on the amount of non-sickling haemoglobin in each RBC, how much of this is due to gene therapy (rather than myeloablation), its durability, and which SCD symptoms or complications are improved by treatment. However, the risks of gene therapy include chemotherapy, the introduction or enhancement of stem cell abnormalities, and possible lack of (long-term) efficacy. Gene addition therapy could result in random vector insertion sites within stem cells, which could potentially lead to cellular proliferation or cancer. Gene editing can also produce unintended modifications, which again could result in cancer. Electroporation could also decrease stem cell survival.58
A Question of Psychological Wellbeing in Sickle Cell Disease: Challenges and Considerations for Best Practice
Kofi A. Anie
SCD can cause a variety of significant challenges, including physical symptoms and psychosocial problems, potentially resulting in a substantial impact on patients and their families.59
The Sickle Cell World Assessment Survey (SWAY) was an international survey that aimed to improve understanding of patient and healthcare provider (HCP) perspectives of living with SCD, with the goal of identifying unmet needs in order to improve quality of life.59 It included 2,145 patients aged ≥6 years, and 365 HCPs from 16 countries.59,60 Overall, 60% of patients reported that SCD had a high impact on their emotional wellbeing, with 58% being frustrated by their symptoms, 58% and 48% being worried that their SCD would worsen or cause them to die, respectively, and 49% feeling stressed due to their SCD (all defined as a score of ≥5 on a Likert scale of 0–7).59 Only 36% reported getting professional emotional support, while 62% wished they could receive such help. Interestingly, HCPs reported a higher emotional impact of SCD on their patients than the patients themselves (impact on emotional wellbeing: 91%; frustrated by symptoms: 82%; worried about worsening or dying: 78% and 70%, respectively; stressed by symptoms: 76%).60
The psychological burden caused by SCD is multifaceted, and can involve symptoms such as perceived stress, depressed mood/anxiety, physiological arousal (e.g., muscle tension, reduced sleep), pain, and negative thoughts.61 External stressors (e.g., job loss, family conflict, bills, childcare, health complications) can also impact the patient. There are various generic and SCD-specific psychological assessments that can be used in patients with SCD, as detailed in Table 1.
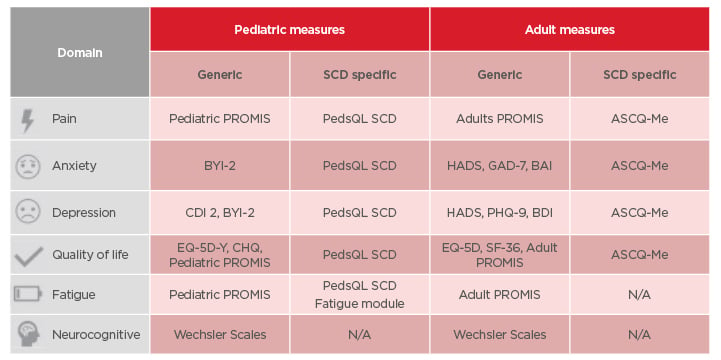
Table 1: Psychological assessments for children and adults with sickle cell disease.
ASCQ-Me: Adult Sickle Cell Quality of Life Measurement Information System; BAI: Beck Anxiety Inventory; BDI: Beck Depression Inventory; BYI-2: Beck Youth Inventory 2nd Edition; CDI 2: Child Depression Inventory 2nd Edition; CHQ: Child Health Questionnaire; EQ-5D: EuroQol-5 Dimension; EQ-5D-Y: EuroQol-5 Dimension (Youth); GAD-7: General Anxiety Disorder-7; HADS: Hospital Anxiety and Depression Scale; N/A: not applicable; PedsQL SCD: Pediatric Quality of Life Inventory Sickle Cell Disease Module; PHQ-9: Patient Health Questionnaire-9; PROMIS: Patient-Reported Outcomes Measurement Information System; SF-36: Short-Form Survey 36.
Psychological interventions can involve addressing pain or fatigue, reducing distress and emotional problems, modifying or enhancing coping ability, addressing misconceptions and stigma, and improving quality of life and emotional wellbeing. They should combine patient assessments with appropriate therapies, such as psychoeducational therapy, behavioural therapy, and supported self-management. Societal education could help patients to feel more comfortable in admitting to having SCD, although younger patients appear to be more confident in their diagnosis, which is perhaps the result of increased awareness on social media. Dedicated psychologists in SCD centres could be beneficial; HCPs could also help patients who suffer with fatigue by informing their employers that they may need to work shorter shifts, for example.
Psychoeducational materials (e.g., books, apps) can improve a patient’s knowledge and understanding of their disease, and support their psychological wellbeing. There are specific materials for children to help them understand their SCD.
CBT is the gold standard of evidence-based interventions.62 This teaches patients how their thoughts, feelings, and behaviours work together, invoking practical methods to solve current problems. Various SCD studies have reported that CBT can reduce healthcare utilisation (in children/adolescents),63 reduce the emotional pain component (in adults),64 reduce anxiety, and improve coping (in adults).65 However, family home-based CBT was found to not be effective in adolescents.66
Mindfulness-based cognitive therapy is a combination of mindfulness techniques (e.g., meditation, breathing, stretching), and elements of cognitive therapy. Acceptance and commitment therapy combines acceptance and mindfulness strategies with commitment and behavioural change to help build resilience. Lastly, motivational interviewing addresses any ambivalence to behavioural change, and provides the motivation to stop bad habits and adhere to medication.
Supported self-management can also be used to empower patients to take control of their own psychological wellbeing. These involve paper materials (e.g., manuals) and mobile health behavioural interventions, such as apps to monitor pain, symptoms, and medication adherence, that can provide patient education, coping skills training, cognitive-guided relaxation, and CBT.67 These apps can be SCD specific (e.g., Sickle-O-Scope), or more generic apps for associated effects such as adherence and depression.
Overall, the psychological wellbeing of patients with SCD is an important issue, and routine assessments are required for timely therapeutic interventions and appropriate support. Fortunately, digital health interventions offer the prospect of real-time self-support.
A Longitudinal Observational Study to Evaluate the Psychometric Properties of a Daily Diary for Adolescents and Adults with Sickle Cell Disease
Robert Clark Brown
Although there are various SCD-specific patient-reported outcome measures to assess the frequency and severity of VOCs and their impact on patients, these require patients to think back to their last VOC and evaluate its severity. They may, therefore, fail to capture the day-to-day impact of VOCs. The SCPD-S app was developed to evaluate the impact of VOCs in adolescents and adults on the days they occur. It is completed daily, and captures VOC duration, healthcare visits, pain severity, use of pain medication, and the impacts of the VOC (missed school/work; interference with school/work, daily living activities, social/recreational activities, sleep; fatigue; emotional difficulty; and difficulty with bathing/showering, indoor mobility, household chores, preparing food). In the absence of a VOC, SCD pain and impacts are recorded instead.
This study will enrol 328 participants with SCD aged ≥12 years who will complete survey questions every day for 90–120 days. Patients will also complete various other assessments during the study.
Factor analysis will be used to examine the conceptual framework of the SCPD-S VOC impact items. This will incorporate multilevel modelling to account for the nested structure of the data. The measurement model will be refined based on the factor analysis results, and the final model will be further evaluated using Mokken scale and multi-trait analyses.
Once the conceptual framework of the SCPD-S impact items has been confirmed, a scoring system will be developed. Three different scoring systems will be examined: daily VOC impact, 7-day VOC impact aggregate, and VOC experience impact aggregate. Planned psychometric analyses include internal consistency reliability, test–retest reliability, convergent and divergent validity, known-groups validity, responsiveness, and differential item functioning.
As of 22nd April 2021, 322 participants had been enrolled. This ongoing study is taking a unique approach to the psychometric validation of a SCD-specific patient reported outcome that was designed to collect daily information on sporadic and unpredictable VOC events. The study procedures promote participant engagement and offer flexibility in completing the daily diary. Once the psychometric properties of the SCPD-S have been documented, the diary could be used in clinical practice to describe patients’ experiences during VOCs and capture responses to treatment.
Low SARS-CoV-2 Seroprevalence in a Cohort of Patients with Sickle Cell Disease: Possible Effects of Emphasis on Social Isolation for a Population Initially Considered to Be at Very High Risk
Patients with SCD were initially considered to be at increased risk of severe COVID-19, but observational studies have indicated that this may not be the case.68-70
In this single-centre Brazilian study, 135 patients with SCD (86% of the total at the centre) were tested for SARS-CoV-2 antibodies from July 2020 to January 2021. Most patients (61%) had HbSS genotype, with 30% having HbSC; 57% were male; and the median age was 42 years in a patient age range of 19 years to 74 years. Most patients (66%) were using hydroxyurea and 4% were on chronic transfusions. During the study period, 45% had VOCs and 27% had symptoms suggestive of COVID-19, but only two had a positive PCR test for SARS-CoV-2.
Overall, 88% of patients considered themselves to be vulnerable to COVID-19 infection, and only 17% believed that individual and collective protection measures were expendable. Most (91%) said that they were able to adopt basic social distancing measures, with 76% cancelling social events, 64% reducing public transport use, and 44% working or studying remotely.
Serological testing was performed once in 58% of patients, twice in 34%, and ≥3 times in 8%. Overall, 15 patients (11%) had positive results. One patient became seropositive during the study period without signs or symptoms of infection, and two patients became seronegative 3 months after a positive test. Seropositivity was associated with older age (p<0.001) and regular public transport use (p=0.02), but not education, income, number of household contacts, or working outside the home. One patient was hospitalised for COVID-19. Despite needing mechanical ventilation and exchange transfusions, they were discharged after 14 days.
Overall, this data shows that social distancing was effective for these patients with SCD, and that patients with SCD do not appear to be at increased risk of severe COVID-19.
Impact of COVID-19 on Sickle Cell Disease in Lebanon: Mild Disease and One Case of Deep Vein Thrombosis
This study included 11 patients, none of whom were of African descent, with SCD who had confirmed COVID-19 in Lebanon. Their median age was 22 years in a range of ages from 14 years to 40 years; nine patients were male; and eight had HbSS. All of the patients were on hydroxyurea (10 of whom were long-term users), one was on voxelotor, one was on crizanlizumab, and none were undergoing chronic transfusions.
Nine patients had moderate-to-severe COVID-19 symptoms, and two had mild symptoms. Seven patients had pain crises; two patients underwent RBC transfusions for pain and anaemia; and one patient had erythrocytapheresis for severe pain. Three patients required oxygen, but none were admitted to a critical care unit. Three patients received corticosteroids, and five received low-molecular-weight heparin.
Although most of these patients with SCD had mild or moderate COVID-19, there were two notable exceptions:
- A 16-year-old male with HbSS genotype developed multisystem inflammatory syndrome and was hospitalised for 26 days. He had prolonged high-grade fever, epigastric pain, and maculopapular rash. He displayed elevated levels of D-dimer (>3000 ng/mL) and received an RBC transfusion, intravenous immune globulin therapy, corticosteroids, low-molecular-weight heparin, and antibiotics.
- An 18-year-old male with HbSS developed extensive deep vein thrombosis in his right leg 20 days after recovering from a very mild COVID-19 infection. This patient had been on aspirin for 6 years after recovering from a catheter-provoked deep vein thrombosis.





