Meeting Summary
Haemophilia A (Factor VIII [FVIII] levels ≤40 IU/dL) is a chronic condition with consequences beyond bleeding complications. Many people with haemophilia A (PwHA) experience pain, joint damage, psychosocial impacts, restrictions in daily activities, and limitations in physical activities. Cédric Hermans, Professor at the Cliniques Universitaires Saint-Luc, Brussels, Belgium, outlined how ambitious treatment goals, beyond converting severe haemophilia A into a more moderate or mild form of the condition, are required. With new treatments, it will be possible to target FVIII activity levels in the non-haemophilia range (>40 IU/dL), allowing PwHA to reach freedom from bleeds, leading to a haemophilia-free mindset, and comparable quality of life (QoL) with their peers. Maria Elisa Mancuso, Senior Haematology Consultant at IRCCS Humanitas Research Hospital, Milan, Italy, highlighted the evolution of haemophilia A treatments; she showed clinical evidence that a zero-bleed goal may require sustained FVIII activity levels >40 IU/dL for complete protection against all types of bleeds and joint damage. Rubén Berrueco, Paediatric Haematologist at the Sant Joan de Déu Barcelona Children’s Hospital, Spain, described the haemophilia paediatric patient journey, and how uncertainties related to bleeds and treatment burden pose unique challenges for children and their caregivers. He presented his perspectives on challenges with current treatments (e.g., delayed inhibitor development, subclinical bleeds, and lack of skills for intravenous administration) and the need to improve self-autonomy and decrease hospital dependency. New treatments to achieve the non-haemophilia range of FVIII could address current unmet needs. The experts discussed that treatments for many diseases (e.g., diabetes, hypertension) aim to restore normal values (blood sugar, blood pressure), which was not the case until now for haemophilia. A more patient-centred approach with treatments targeting normal values of FVIII could allow all PwHA to become mentally and physically liberated from the constraints of their condition, and to live with optimised health and well-being.
![]()
Erratum: Normalisation of Haemostasis in Haemophilia A
Symposium chairperson: Cédric Hermans
Symposium speakers: Maria Elisa Mancuso and Rubén Berrueco
Original citation: EMJ Hematol. 2024;12[1]:29-37. https://doi.org/10.33590/emjhematol/TUPO1598.
Date correction published: 08.08.2024
In the article reviewing a satellite symposium session presented at the EHA 2024 Hybrid Congress by Cédric Hermans, Maria Elisa Mancuso, and Rubén Berrueco, published in EMJ Hematology 12.1 on the 25th of July 2024, the following sentence on page 31 is incorrect: ‘non-factor therapies target an equivalent homeostatic activity in the mild haemophilia range, with the aim of providing improved protection’. This sentence should instead read: ‘non-factor therapies target an equivalent haemostatic activity in the mild haemophilia range, with the aim of providing improved protection’. This has now been amended.
EMJ apologises for the error and any inconvenience caused.
![]()
Click here to view the highlights
Welcome and Introduction
Cédric Hermans
Haemophilia A is an inherited deficiency in coagulation FVIII. Based on their residual FVIII activity levels, PwHA are considered to have either severe (FVIII <1 IU/dL), moderate (FVIII 1–5 IU/dL), or mild (FVIII >5–40 IU/dL) disease.1 People with mild disease tend to bleed only after trauma, whereas those with severe or moderate disease also experience spontaneous bleeds into joints and muscles.1,2 However, residual FVIII activity levels do not always correlate with bleeding manifestations, and people with moderate or mild disease can have a similar bleeding phenotype to that associated with severe haemophilia A.3 The World Federation of Hemophilia (WFH) currently recommends regular replacement therapy (prophylaxis) with clotting factor concentrates or other haemostasis products for all PwH with a severe bleeding phenotype, regardless of their laboratory-assigned severity, with a recommended FVIII target trough level of >3–5 IU/dL or higher.1 Hermans explained how the consequences of haemophilia go beyond bleeding complications, with many PwHA experiencing joint damage, acute and chronic pain, limitations in physical activities, restrictions in daily lives, and psychosocial impacts (Figure 1).4-9
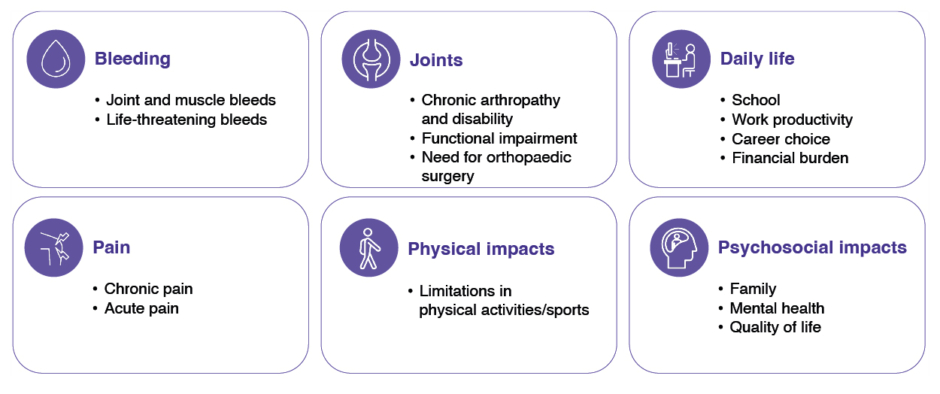
Figure 1: Consequences of haemophilia: more than bleeds.4-9
In the past, haemophilia treatment aimed to convert severe disease into a moderate form (FVIII levels around 1–2%) to prevent life-threatening bleeds; however, this is not sufficient to protect all PwHA from bleeds and specifically joint damage.10 More sophisticated treatment options, such as extended half-life (EHL) FVIII products are maintaining FVIII levels around 3-5% (FVIII trough levels in moderate range and FVIII peak levels in non-haemophilia range), and non-factor therapies target an equivalent haemostatic activity in the mild haemophilia range, with the aim of providing improved protection.11-13 Now for the first time, with gene therapy and ultra-long FVIII, it is feasible to reach a new ambitious goal for PwHA: the normalisation of haemostasis (i.e., maintaining FVIII activity levels in the non-haemophilia range [>40 IU/dL]) without increasing treatment burden for PwHA (Figure 2).14-16
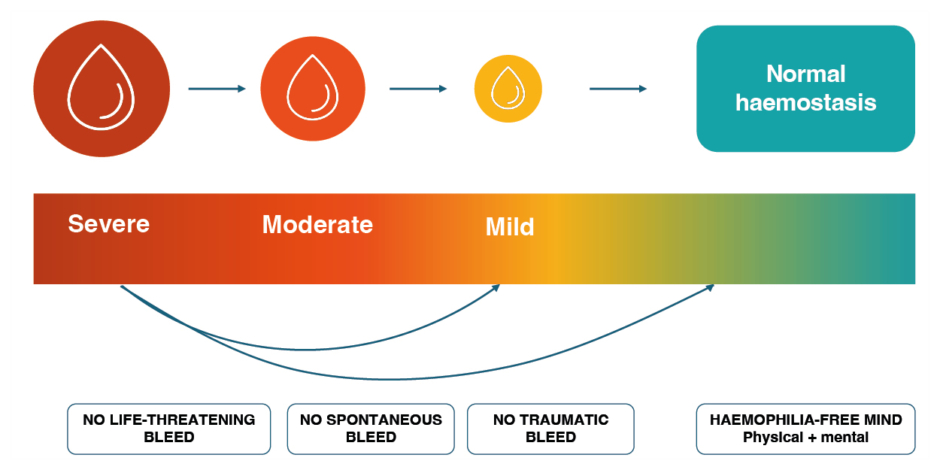
Figure 2: The evolving goals of haemophilia therapy: from saving life to normalising life.16-18
As haemophilia therapies are evolving, the expectations and aspirations of PwHA have moved beyond controlling symptomatic bleeds towards mental and physical liberation from the constraints of haemophilia and its treatment.16,18 By achieving sustained FVIII levels in the non-haemophilia range, health equity is becoming a realistic possibility for PwHA.16
Expanding Possibilities for People with Haemophilia A
Maria Elisa Mancuso
Mancuso acknowledged the tremendous success in the development of haemophilia therapies over the past five decades. Each innovation stemmed from the design of new molecules that addressed different unmet needs and allowed for the inclusion of new outcome measures. These ranged from preventing death, preventing joint disease, and improving QoL and other patient-reported outcomes, to targeting the non-haemophilia range of FVIII and heading towards health equity with people who do not have haemophilia.16,19-21 Prophylaxis is recognised as the standard of care for people with haemophilia and a severe bleeding phenotype; 1 however, to optimise a treatment regimen, a dynamic and patient-centric approach is needed, taking individual needs into account.22 Evidence is emerging that the guideline-recommended FVIII trough levels of 3–5 IU/dL are not enough to prevent subclinical bleeding and joint damage in all patients, and that for a zero joint bleed goal (including silent bleeds), FVIII levels in the non-haemophilia range may be required.23 Results from the Phase III GENEr8-1 trial with the gene therapy valoctocogene roxaparvovec in haemophilia A showed that, overall, good bleed control was achieved in the majority of participants, but only people with FVIII levels >40 IU/dL were 100% bleed-free.24 However, FVIII activity levels achieved with valoctocogene roxaparvovec declined over time, with 10.6% of patients maintaining levels in the non-haemophilia range 3 years after infusion.15 Non-factor replacement therapies such as emicizumab, fitusiran, and anti-tissue factor pathway inhibitor agents (concizumab and marstacimab) generate peak-and-trough-free steady states that likely achieve a correspondence to coagulation activation in the range of mild haemophilia.25 The ultra-long FVIII product efanesoctocog alfa has a three-fold longer half-life compared to conventional EHL FVIII products,26 allowing maintenance of FVIII levels >40 IU/dL with a once-weekly dose of 50 IU/kg for up to 4 days,14, 26 thereby providing significant bleed protection with reduced treatment burden for PwHA.14
People with non-severe haemophilia are not uniformly protected from the development of arthropathy, with a proportion requiring prophylaxis.3 Mancuso mentioned that, to protect all PwHA from arthropathy, it is necessary to look beyond annualised bleeding rates (ABR) and employ imaging techniques such as ultrasound and MRI to monitor joints for subclinical bleeding and early evidence of joint damage.27,28 In a Dutch study of 43 people with severe haemophilia A, MRI detected haemosiderin deposits in 16% of all screened people; of these, 43% had synovial hypertrophy and/or osteochondral changes.29 Without regular monitoring, synovial proliferation may be overlooked, as demonstrated in a recent ultrasound study of 79 people with severe haemophilia A without recent joint bleeds.30 In this cohort, ultrasound detected active synovial proliferation in 22% of patients; of these, 82% had no clinical signs of inflammation (swelling or warmth).30
Synovitis is the first step towards irreversible joint damage, but it can be reversed by intensified prophylaxis with FVIII and anti-inflammatory drugs, as recognised by the guidelines of the German Thrombosis Society.1,31,32 These guidelines recommend 1–2 daily doses of 40–60 IU/kg coagulation factor initially for acute synovitis, and a 6-month trough level target of ≥30 IU/dL to treat chronic synovitis (Figure 3).32
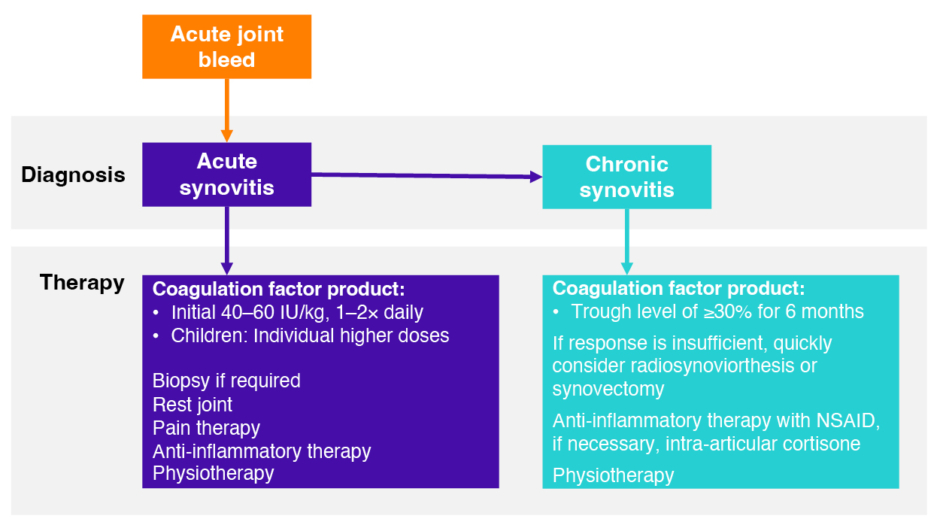
Figure 3: German Thrombosis Society treatment algorithm for synovitis in haemophilia.32
NSAID: non-steroidal anti-inflammatory drug.
Patient Case 1
Mancuso presented the case of ‘Paul’, a 34-year-old physically active person who has moderate haemophilia with no history of inhibitors (neutralising antibodies against FVIII concentrates) and a baseline FVIII activity level of 2.3 IU/dL. ‘Paul’ works, travels often, and participates in sports on a regular basis. Secondary prophylaxis was started with thrice-weekly standard half-life recombinant FVIII at 4 years of age before switching to EHL FVIII every fifth day in 2016 due to time constraints impacting on the ability to perform thrice-weekly infusions. The patient started to experience ankle pain, especially in the morning, and minor bleeding, and the prophylaxis schedule was intensified to a twice-weekly infusion of 45 IU/kg. In their joint evaluation, a haemophilia joint health score of 6, and a haemophilia early arthropathy detection with ultrasound score of 4 were noted; the ultrasound investigation found evidence of synovitis in their right ankle that had not presented 12 months previously. The prophylaxis intensification was discussed with ‘Paul’, whose priorities were to stay active and independent, and be free of pain whilst maintaining a feasible treatment schedule. Mancuso highlighted the importance of discussing the clinical meaning of synovitis and the need for regular joint monitoring with patients, along with giving advice on how to manage synovitis with physiotherapy and anti-inflammatory medication in addition to high-factor treatment, whilst keeping an open mind regarding treatment and schedule changes.
Realising New Opportunities in Children
Rubén Berrueco
Berrueco described the human-centred process carried out at his centre, aimed to better understand the unmet needs of children with haemophilia A (CwHA) and their caregivers. This qualitative research consisted of semi-structured interviews with healthcare professionals (HCP), CwH, and their caregivers, and other stakeholders, as well as observation during clinical appointments at the centre. The information allowed them to draw the paediatric haemophilia journey and to describe the unmet needs that CwH experience through time. Potential moments to improve CwH’s and their caregivers’ experience are (i) treatment initiation, when caregivers tend to be keen to learn about haemophilia but, at the same time, experience many uncertainties related to the condition, and (ii) when CwH are 7–8 years old, and first notice that they are different to their peers and begin to ask, “why me?”
Apart from the unmet needs described by the patients, Berrueco believes that it is important to explain to parents that, even with treatment, HCPs are aware of other unmet needs that must be addressed. Haemophilia A is a chronic disease with a high treatment burden that impacts on daily activities and QoL.33 Moreover, CwHA are still at risk of life-threatening bleeds such as intracranial haemorrhage,34 a risk that can be significantly mitigated by prophylaxis.35 Importantly, compared with on-demand treatment, prophylaxis with FVIII is associated with a lower rate of inhibitors,36 and early primary prophylaxis with FVIII has been related to better joint health results.37,38 However, the Joint Outcome Study and subsequent Joint Outcome Continuation Study demonstrated that, despite prophylaxis with FVIII, joint damage can also occur in the absence of recognised bleeds.28 Nowadays, many CwHA are treated with non-factor treatment (emicizumab), but it is important to highlight that there is a paucity of data for this treatment regarding the risk of inhibitor development following on-demand FVIII exposure,39,40 and regarding the predictability of joint health.41
For prophylaxis to be successful, adherence is crucial at any age, but can be problematic in adolescents and young adults.42,43 Adolescents often consider prophylaxis infusions a time-consuming and inconvenient interference with their daily lives. They may lack the necessary skills to self-infuse, be phobic of needles, be forgetful, or lack family support.44,45 Educating adolescents about the consequences of non-adherence is crucial, and self-injection should be encouraged as early as possible in the patient journey to ensure young people with haemophilia gain autonomy and good self-management skills.43,44
Berrueco discussed how sports participation is an important way to improve QoL in CwHA;46 at the same time, this needs to be balanced with the associated increased bleed risk.46 Achieving normalisation of haemostasis and a ‘haemophilia-free mind’ could enable CwHA to live full lives with the same aspirations as their peers without haemophilia.47 In addition to considering the unmet needs in CwHA (Figure 4), Berrueco urged HCPs not to forget that caregivers continue to need support through all stages of their child’s development.
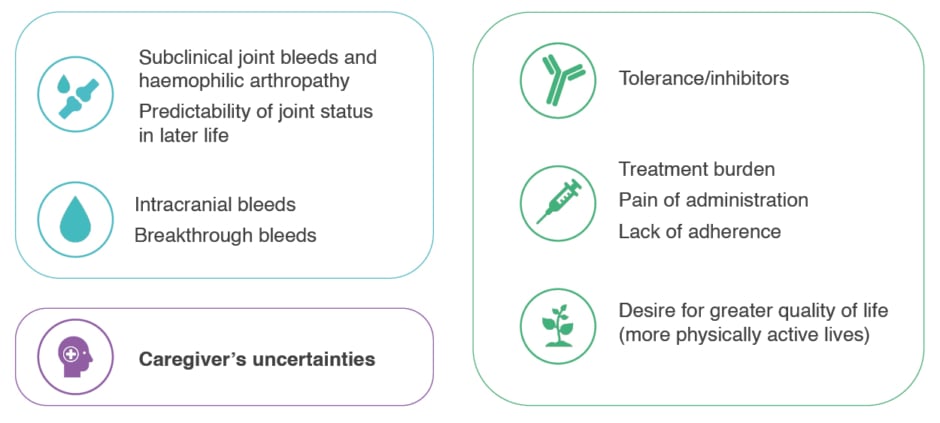
Figure 4: Many unmet needs still exist for paediatric patients with haemophilia A.28,41,42,48,49
Treatment of CwHA has changed over the years, and tremendous progress has been achieved.21 A decade ago, FVIII prophylaxis was started at around 2 years of age, with a twice-to-three-times weekly infusion regimen,50 and tolerisation was achieved within 6 months.41 HCP concerns were focused on the type of FVIII product they should administrate. Now, prophylaxis is started even earlier, and worries are related to the protection against subclinical bleeds and the potentially increased risk to develop inhibitors at later age.
Recent and new treatment approaches offer opportunities to address unmet needs in CwHA. Emicizumab allows for either once-weekly, every 2-, or every 4-week subcutaneous injections,39,51 achieving FVIII equivalence of between 9–20 IU/dL.52-55 Whilst the recently published HAVEN 7 study of emicizumab in infants with severe haemophilia A showed no new safety signals in this age group, with a model-based ABR of 0.4 and 54.5% of participants reaching zero treated bleeds,51 real-world data in 314 young people with severe haemophilia A with and without inhibitors found that 15 participants experienced at least one severe muscle bleed.56 The pharmacokinetic profile of the ultra-long FVIII efanesoctocog alfa supports a once-weekly dosing schedule, maintaining FVIII in the non-haemophilia range for up to 3 days in children.57 In the Phase III XTEND-Kids study in boys younger than 12 years with severe haemophilia who had previously been treated with FVIII, the estimated mean ABR in the sensitivity population (n=73) was 0.61, and 64% of patients reached zero bleeds;58 crucially, no inhibitors were detected.59
Patient Case 2
Berrueco presented the case of ‘Valentín’, a 7-year-old boy with severe haemophilia A who started emicizumab at the age of 3 years (after >50 ED with FVIII), and recently started playing soccer. In a recent evaluation of the patient’s joint health, there was evidence of right knee swelling and a new synovitis in the suprapatellar recess. During further discussion with the parents, it was remembered that ‘Valentín’ had experienced knee pain for the past few weeks, suggesting that the synovitis was active (symptomatic). The clinical goal for this child was to achieve and then maintain optimal joint health, whilst allowing him to have a QoL of life comparable with his peers. For ‘Valentín’ and his parents, maintaining physical activity and having a low treatment burden were priorities. It was agreed that close clinical monitoring was necessary, and that the treatment schedule and other treatment options should be reviewed to achieve the desired clinical goal for this child and his caregivers.
Panel Discussion and Q&A: Resetting Goals and Approaches in Haemophilia A
All Faculty
Speaker presentations were followed by a combined panel discussion and audience Q&A session. Mancuso explained that, to date, no treatment for haemophilia A is curative, but if joint health is already poor, a higher level of protection is required. Hermans noted that, for other acquired conditions (e.g., hypertension, diabetes, hypercholesterinaemia), the treatment goal is to restore normal physical or biological parameters;16 in haemophilia A, the treatment goal of trough levels of 3–5% merely converts severe-to-moderate disease, which Hermans argued is insufficiently ambitious. The goal of normalisation has simply not been considered achievable or affordable, but with new treatment options, it is now becoming realisable.16 Berrueco stressed the importance of looking beyond the impact of haemophilia on children and their parents, and also consider other family members, teachers, and healthcare providers. It was agreed that families need to be continuously educated on the treatment options and long-term treatment effects. With adequate monitoring, it is expected that sustaining FVIII activity levels in the non-haemophilia range will protect joint health and thereby reduce sequelae of haemophilia later in life; monitoring with ultrasound also increases patient education and adherence. In adult PwHA, FVIII activity levels in the non-haemophilia range can offer a level of protection that allows treatment for comorbidities, such as cardiovascular pathologies, without having to prioritise bleed prevention or prevention of thrombotic events. The ‘haemophilia-free mind’ is an aspirational concept, and with current treatments, PwHA will still have a life-long serious disease,19 but a reduction of the burden of treatment and better haemostatic protection can contribute to alleviating the burden of haemophilia. All panel members agreed that clinical studies should address outcomes beyond the prevention of bleeds, and include a focus on patient-related outcomes.60





